FACOLTÀ DI SCIENZE MM.FF.NN.
CORSO DI LAUREA IN BIOTECNOLOGIE AGROINDUSTRIALI
TESI DI LAUREA
STUDIO DI MODELLI DI PROTEINE ANTENNA DI PIANTE MEDIANTE SIMULAZIONE MOLECOLARE AL CALCOLATORE
Relatore: Prof. HENRIETTE MOLINARI
Correlatori: Dott. FEDERICO FOGOLARI
Dott. MASSIMO CRIMI
Laureando: GIUSEPPE INSANA
--------------------------
ANNO ACCADEMICO 1998-99
Ringraziamenti
il Dott. Federico Fogolari, per la sua attiva collaborazione in ogni fase del lavoro di tesi;
il Dott. Massimo Crimi, per la disponibilità ed attenzione con cui mi ha seguito e per i suoi preziosi consigli.
Sommario
A. SCOPO DELLA TESI *
B. INTRODUZIONE *
II. LA FOTOSINTESI *
II.1 GENERALITÀ *
II.2 I CLOROPLASTI *
II.3 INTERAZIONE LUCE-MATERIA *
II.4 I PIGMENTI FOTOSINTETICI *
II.5 INTRODUZIONE AI SISTEMI ANTENNA *
II.6 LE PROTEINE ANTENNA DEL PS II *
II.7 NOMENCLATURA PROTEINE CAB *
III. EVOLUZIONE PROTEICA *
IV. OMOLOGIE ED ANALOGIE *
V. BIOINFORMATICA *
V.1 DATABASE PUBBLICI *
V.2 ALLINEAMENTI DI SEQUENZE *
V.3 PREDIZIONE DELLA STRUTTURA PROTEICA *
VI. MODELLISTICA MOLECOLARE *
VI.1 GENERALITÀ *
VI.2 CAMPI DI FORZE *
VI.3 MINIMIZZAZIONE E DINAMICA *
VI.4 ORIENTARE UNA SIMULAZIONE *
VII. LA COVARIANZA *
VIII. ELEMENTI CHIAVE DI STRUTTURA *
VIII.1 PONTI SALINI *
VIII.2 PONTI IDROGENO *
VIII.3 a -ELICHE *
I.1 Insight *
I.2 Discover *
I.3 WhatIf *
I.4 Swiss-PDB Viewer *
I.5 Gast-Mars *
I.6 Maxsprout *
I.7 HIC-Up *
II. ALLINEAMENTO, PREDIZIONE ED ANALISI DI SEQUENZA *
II.1 Database Di Sequenze *
II.2 ClustalW *
II.3 Macaw *
II.4 Predict Protein *
II.5 Phylip *
II.6 AliAna *
II. RICOSTRUZIONE STRUTTURA (Catene laterali e cromofori) *
III. MODIFICHE APPORTATE AL CAMPO DI FORZE CVFF PER LA PARAMETRIZZAZIONE DEL MAGNESIO *
III.1 DISTRIBUZIONE DELLA CARICA *
III.2 DEFINIZIONE DI NUOVI TIPI ATOMICI *
IV. DINAMICA MOLECOLARE *
V. LA FAMIGLIA MULTIGENICA *
V.1 ALLINEAMENTO TRA LHC II E LE ANTENNE MINORI *
V.2 ESTENSIONE DELL'ALLINEAMENTO *
V.3 RAPPRESENTAZIONE AD ALBERO FILOGENETICO *
VI. CONFRONTI DI SEQUENZA *
VI.1 STUDI DI COVARIANZA *
VII. IL TRIMERO *
VIII. IPOTESI DI MODELLO *
VIII.1 MODELLO 1 (PONTI SALINI: E139-R142, E180-R70, E63-K177) *
VIII.2 MODELLO 2 (PONTI SALINI: E139-R70, E63-R142, E180-K177) *
VIII.3 MODELLO 3 (PONTI SALINI: E63-R142, E180-R70) *
VIII.4 CONCLUSIONI *
IX. MODELLISTICA PER OMOLOGIA DELLE ANTENNE MINORI *
IX.1 CP 29 *
IX.2 CP 26 *
IX.3 CP 24 *
F. APPENDICI *
II. TABELLA DI RIFERIMENTO NUMERAZIONE AMINOACIDICA *
III. PARAMETRI INERENTI ALLE SIMULAZIONI MOLECOLARI *
IV. ANALISI DISTORSIONI NELLA STRUTTURA TRIDIMENSIONALE *
Indice delle Figure
Figura B-1: Il cloroplasto e la sua struttura *
Figura B-2: Schema rappresentante la membrana tilacoidale ed i complessi coinvolti nel trasporto elettronico *
Figura B-3: Meccanismi di eccitazione e diseccitazione *
Figura B-4: La molecola di clorofilla *
Figura B-5: Schema a Z di Bendall e Hill. In ordinata il livello di potenziale redox *
Figura B-6: Struttura di alcuni carotenoidi *
Figura B-7: Rappresentazioni della struttura cristallografica di proteine antenna procariotiche *
Figura B-8: Arrangiamento del PS II proposto da Kilian et al. [1998] *
Figura B-9: Rappresentazione schematica della disposizione e coordinazione dei pigmenti in LHC II *
Figura B-10: Rappresentazione della struttura di LHC II *
Figura B-11: Energia e probabilità di una particella classica e quantomeccanica in un oscillatore armonico *
Figura B-12: Linee di ricerca del minimo per una superficie bidimensionale di energia *
Figura C-1: Visualizzazione grafica dei termini dell?equazione adottata da CVFF *
Figura C-2: Esempio di applicazione della maschera strutturale nella ricerca di possibili ponti salini *
Figura D-1: Struttura delle clorofille inserite nella struttura ricostruita *
Figura D-2: Rappresentazione schematica della struttura degli atomi Mg e N nella clorofilla *
Figura D-3: Diversità nella distribuzione di carica data dagli algoritmi di Insight e Gast-Mars *
Figura D-4: Distribuzione di carica adottata per le clorofille della struttura ricostruita *
Figura D-5: Istogramma di distribuzione relativo alle lunghezze del legame Mg-N *
Figura D-6: Rappresentazione dell'angolo N-Mg-N *
Figura D-7: Istogramma relativo all'angolo N-Mg-N *
Figura D-8: Rappresentazione dell'angolo N-Mg-N *
Figura D-9: Istogramma relativo all'angolo N-Mg-N *
Figura D-10: Rappresentazione dell'angolo Mg-N-C *
Figura D-11: Istogramma relativo all'angolo Mg-N-C *
Figura D-12: Rappresentazione dell'angolo C-N-C *
Figura D-13: Istogramma relativo all'angolo C-N-C *
Figura D-14: Istogrammi relativi a tutte le clorofille con sovrapposte le Gaussiane approssimanti *
Figura D-15: Variazione delle costanti energetiche nel corso della simulazione *
Figura D-16: Variazione delle costanti energetiche per i vincoli imposti nel corso della simulazione *
Figura D-17: Grafico rappresentante l'andamento delle energie durante il simulated annealing *
Figura D-18: Grafico rappresentante l'andamento delle temperature durante il simulated annealing *
Figura D-19: Rappresentazione a fotogrammi della simulazione del protocollo a costanti di forza rilassate *
Figura D-20: Esemplificazione grafica del risultato del protocollo approntato *
Figura D-21: Rappresentazione a blocchi di omologia dell'allineamento risultante *
Figura D-22: Rappresentazione ad albero della distanza tra le sequenze proteiche nella famiglia Lhcb *
Figura D-23: Ricorrenza aminoacidica e sequenza consenso per la zona transmembrana *
Figura D-24: Forma grafica delle matrici a punti create in seguito all'analisi del programma AliAna *
Figura D-25: Analisi di covarianza per residui ionizzabili sulla famiglia Lhcb *
Figura D-26: Analisi di covarianza su un sottoinsieme rappresentativo delle proteine Lhcb *
Figura D-27: Analisi di covarianza per residui ionizzabili sulle proteine LHC II di tipo I e II *
Figura D-28: Analisi dei possibili ponti salini in LHC II tipo I *
Figura D-29: Andamento delle energie nel corso della simulazione del trimero *
Figura D-30: Andamento delle temperature nel corso della simulazione del trimero *
Figura D-31: Rappresentazioni della struttura ricostruita del trimero *
Figura D-32: Ricorrenza aminoacidica e sequenza consenso N-terminale per LHC II tipo I e II *
Figura D-33: Ricorrenza aminoacidica e sequenza consenso C-terminale per LHC II tipo I e II *
Figura D-34: Rappresentazione della struttura dei due ponti salini complessi ipotizzati *
Figura D-35: Rappresentazione del modello 1 (in alto senza ed in basso con le clorofille) *
Figura D-36: Rappresentazione del modello 2 (in alto senza ed in basso con le clorofille) *
Figura D-37: Rappresentazione del modello 3 (in alto senza ed in basso con le clorofille) *
Figura D-38: Rappresentazione dell'ipotizzata coordinazione della clorofilla a3 in CP 24 *
Nel lavoro di tesi si presenta uno studio di modellistica molecolare per proteine antenna di piante superiori basato sulle informazioni ottenibili dalle sequenze della famiglia multigenica Lhcb (proteine antenna del fotosistema II) e dalla struttura cristallografica della proteina LHC II (Light-Harvesting Complex, complesso di raccolta della luce del fotosistema II) determinata, per quanto riguarda la posizione dei carboni alfa delle eliche transmembrana, mediante microscopia elettronica ad una risoluzione di 3.4 Å [Kühlbrandt et al. 1994, Nature 367: 614-621].
L'importanza di questa struttura risiede nell'essere la prima, e a tutt'oggi l'unica, struttura di proteina antenna risolta di pianta superiore. Inoltre, LHC II è la proteina di membrana più abbondante nei cloroplasti (organelli fotosintetici delle cellule vegetali) coordinando la metà dei pigmenti coinvolti nella fotosintesi delle piante.
I pigmenti (clorofille e carotenoidi) sono legati in modo non covalente alla struttura polipeptidica e sono responsabili della cattura dell'energia luminosa e del trasferimento della stessa al centro di reazione - dove avvengono le reazioni fotosintetiche - nonché della protezione del sistema dalla foto-ossidazione.
L'approccio computazionale e bioinformatico consente di costruire modelli conformazionali per le catene laterali non risolte nella struttura cristallografica e di eseguire simulazioni molecolari su questi modelli.
L'alta omologia delle sequenze disponibili (depositate in banche dati pubbliche) rende possibile lo studio comparato delle sequenze (fornendo ulteriori informazioni strutturali) e di proporre modelli per omologia per le proteine (antenne minori) omologhe a LHC II.
In particolare il lavoro di tesi consiste in:
La determinazione della struttura cristallografica
di LHC II ottenuta per microscopia elettronica su cristalli bidimensionali
ad una risoluzione di 3.4 Å [Kühlbrandt et al. 1994] rappresenta
una chiave fondamentale per la comprensione del funzionamento e dell?organizzazione
di una classe importantissima di proteine delle piante superiori: le proteine
antenna (light-harvesting), responsabili di gran parte dell?assorbimento
dell?energia luminosa, del trasferimento di questa sotto forma di energia
di eccitazione e dei fenomeni di fotoprotezione.
Questa struttura infatti mostra che i sistemi antenna presenti nelle piante superiori sembrano avvalersi di approcci diversi nell?organizzazione strutturale dei pigmenti di clorofilla rispetto ai corrispettivi batterici (soprattutto in relazione alla coordinazione delle clorofille che nelle strutture procariotiche note avviene solo con residui di Istidina al contrario di LHC II in cui altri residui sembrano essere coinvolti), offrendo spunti per ulteriori studi e sperimentazioni.
Questo lavoro di tesi di laurea si concentra sulla possibilità di studiare una proteina la cui struttura sia stata solo parzialmente risolta ed estrarre maggiori informazioni sulla sua struttura e sulla struttura di proteine ad essa omologhe mediante l'uso di strumenti informatici, ovvero delle possibilità di simulazione e di calcolo offerte da un moderno elaboratore.
L'approccio bioinformatico adoperato consiste nello sfruttare tutte le possibili informazioni sia a livello di sequenza che a livello di struttura per costruire un modello, ovvero una rappresentazione che si avvicini il più possibile alla realtà e permetta di tracciare ipotesi da verificare sperimentalmente.
In questa tesi sono stati utilizzati e proposti strumenti e protocolli bioinformatici che hanno permesso di approfondire le conoscenze tridimensionali (tramite studi di sequenza) della proteina LHC II, di ricostruirne la struttura proponendo una collocazione plausibile per le catene laterali (non visibili nella struttura cristallografica) e di fornire diverse ipotesi di modello per residui chiave per il mantenimento della struttura e della funzione offrendo la possibilità di elucidare i motivi strutturali alla base delle diverse funzioni svolte dalle proteine appartenenti alla stessa famiglia.
Nel corso della tesi verranno usate le seguenti abbreviazioni:
| ATP: Adenosine Triphosphate | Chl: Chlorophyll (clorofilla) |
| DNA: DeoxyriboNucleic Acid (acido deossiribonucleico) | DSSP: Definition of Secondary Structure of Proteins |
| EBI: European Bioinformatics Institute | EMBL: European Molecular Biology Laboratory (Heidelberg, Germany) |
| FSSP: Families of Structurally Similar Proteins | FTP: File Transfer Protocol (protocollo di trasferimento archivi) |
| HSSP: Homology-derived Secondary Structure of Proteins | LHC: Light Harvesting Complex (complesso di raccolta della luce, sistema antenna) |
| NAD: Nicotinamide Adenine Dinucleotide | NCBI: National Center for Biotechnology Information |
| NMR: Nuclear Magnetic Resonance (risonanza magnetica nucleare) | NOE: Nuclear Overhauser Effect |
| PDB: Protein Data Bank | PIR: Protein Identification Resource database |
| PS: PhotoSystem (fotosistema) | RNA: RiboNucleic Acid (acido ribonucleico) |
| SDS-PAGE: Sodium Dodecyl Sulphate-PolyAcrylamide Gel Electrophoresis | WWW: World Wide Web (rete multimediale) |
Inoltre compaiono le comuni abbreviazioni ad una e a tre lettere per gli aminoacidi.
Sono quindi riassunte nella seguente tabella:
| A | Ala | Alanina | M | Met | Metionina |
| C | Cys | Cisteina | N | Asn | Asparagina |
| D | Asp | Acido Aspartico | P | Pro | Prolina |
| E | Glu | Acido Glutammico | Q | Gln | Glutammina |
| F | Phe | Fenilalanina | R | Arg | Arginina |
| G | Gly | Glicina | S | Ser | Serina |
| H | His | Istidina | T | Thr | Treonina |
| I | Ile | Isoleucina | V | Val | Valina |
| K | Lys | Lisina | W | Trp | Triptofano |
| L | Leu | Leucina | Y | Tyr | Tirosina |
Vengono utilizzati i simboli chimici per indicare gli atomi:
| C: Carbonio | Ca: Calcio |
| H: Idrogeno | Mg: Magnesio |
| N: Azoto | O: Ossigeno |
La fotosintesi è il processo di trasformazione dell'energia
elettromagnetica della luce in energia chimica. Tale conversione è
il primo passo nella fissazione del carbonio in composti organici a partire
da composti inorganici elementari tramite reazioni ossido-riduttive.
La fotosintesi ossigenica - propria dei piante superiori, cianobatteri, alghe, diatomee, crisofite e dinoflagellati - utilizza acqua come donatore di elettroni e anidride carbonica come accettore secondo la reazione complessiva:
6 H2O + 6 CO2 + luce ® C6H12O6 + 6 O2
Tale processo è costituito da due fasi, classicamente definite fase luminosa e fase oscura, per distinguere le reazioni in base alla loro dipendenza dalla luce.
La fase luminosa consiste nella cattura dell'energia luminosa da parte di complessi proteici contenenti pigmenti antenna il cui stato elettronico eccitato viene trasferito al centro di reazione dove avvengono le reazioni fotochimiche (questo processo è definito light-harvesting ovvero raccolta, cattura della luce):
2 NADP+ + 2 H2O + luce ® 2 NADPH + O2 +2 H+ [1]
ADP + Pi + 3 H+ ® ATP [2]
La reazione [2] è denominata fotofosforilazione e comporta la sintesi di ATP sfruttando un gradiente protonico transmembrana generato in seguito alla reazione [1].
Nella fase luminosa vengono trasferiti elettroni contro gradiente elettrochimico sfruttando l'energia fotonica. Si ha quindi l'accumulo di potere riducente (NADPH) e energia chimica (ATP) (cfr. § B.II.4.1.2) che vengono utilizzati nella fase oscura per la riduzione della CO2 (fissazione della CO2) e produzione di carboidrati:
6 CO2 + 12 H2O + 12 NADPH + 18 ATP ® C6H12O6 + NADP+ + 6 H+ + 18 ADP + 18 Pi
Negli eucarioti (alghe e piante superiori) i processi fotosintetici
avvengono nel cloroplasto, un organello subcellulare specializzato.
I cloroplasti si trovano principalmente nelle cellule del mesofillo, hanno forma elissoidale con una lunghezza di circa 5 m m.
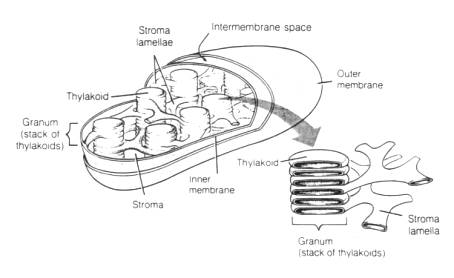
Sono delimitati da una doppia membrana di rivestimento (detta envelope) che racchiude una matrice acquosa - detta stroma - e le membrane interne fotosintetiche - dette tilacoidi.
La principale funzione del rivestimento è di controllare e regolare il movimento di metaboliti, lipidi e proteine in entrata e in uscita dal cloroplasto. La membrana più esterna è altamente permeabile mentre quella interna contiene specifici trasportatori.
Lo stroma contiene il DNA, l'RNA e i ribosomi che consentono al cloroplasto la sintesi autonoma di alcune proteine. Altre proteine necessarie al funzionamento dell'apparato fotosintetico vengono sintetizzate nel citoplasma e importate all'interno del cloroplasto attraverso l'envelope.
Nello stroma avvengono le reazioni della fase oscura ed in esso sono localizzati gli enzimi necessari, in particolare la ribulosio bisfosfato carbossilasi (rubisco), l'enzima responsabile della fissazione di CO2 atmosferica in composti organici.
Le membrane tilacoidali formano un intreccio tridimensionale continuo e chiuso che delimita un secondo compartimento: il lumen tilacoidale.
Queste membrane sono distinte in due tipi di domini: quelle impilate una sull'altra (regioni appressate) a formare strutture dette grana e quelle che rimangono non impilate, lamelle singole che interconnettono diversi grana: regioni non appressate altrimenti dette lamelle stromatiche.
I complessi proteici partecipanti alla fase luminosa della fotosintesi sono inseriti nella membrana tilacoidale: i due fotosistemi (PS I e PS II) con le loro proteine antenna, il complesso del citocromo b6f e l'ATP sintasi.
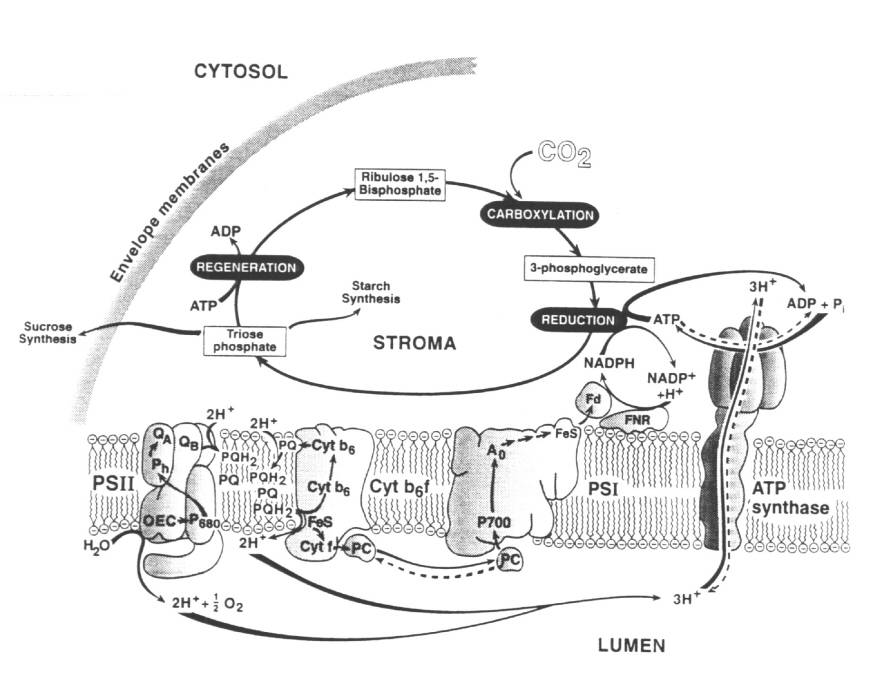
Figura -2: Schema rappresentante la membrana tilacoidale ed i complessi coinvolti nel trasporto elettronico
La lunghezza d'onda (l ) associata ad una particella di massa m e velocità v è data dalla relazione di De Broglie:
l =h/mv
con h costante di Planck (6.626 × 10-34 J× s).
La luce è una radiazione elettromagnetica con l e n (frequenza) legate dall'equazione:
n =c/l
dove c è la velocità (costante) della luce nel vuoto (299800 km/s).
La luce possiede anche una natura corpuscolare, propagandosi sotto forma di particelle chiamate fotoni. L'energia luminosa è quantizzata ovvero si trasmette in pacchetti discreti di energia chiamati quanti piuttosto che con continuità.
L'energia associata ad ogni fotone, il quanto, è calcolabile secondo la relazione:
E=hn
(fotoni di luce con lunghezza d'onda minore trasportano energia maggiore).
Anche l'energia degli atomi è quantizzata, ovvero ogni atomo possiede dei livelli energetici discreti. A ciascuno di essi corrisponde una diversa distribuzione statistica degli elettroni attorno al nucleo nei diversi orbitali atomici. Un orbitale atomico è definito da una funzione d'onda che dà la probabilità di trovare l'elettrone in una regione definita dello spazio. Solitamente si intende orbitale la regione entro cui la probabilità di trovare l'elettrone è superiore al 90%.
Nelle molecole gli elettroni non sono confinati ai singoli atomi ma si muovono in traiettorie che vengono definite orbitali molecolari. La rappresentazione è infatti quella della combinazione di n orbitali atomici a formare n orbitali molecolari.
Ogni stato quantico elettronico possiede dei sottostati vibrazionali e rotazionali con energie lievemente diverse. Vi sono quindi bande di lunghezze d'onda (invece che singole lunghezze d'onda) rappresentanti i diversi livelli.
Gli elettroni sono in grado di interagire con la luce ed essere "promossi" ovvero passare da un orbitale molecolare a minor energia (stato fondamentale, SF) ad uno ad energia più elevata (stato eccitato, SE). Questa transizione si verifica solamente nel caso che il fotone possieda un'energia corrispondente alla differenza energetica tra i due stati.
Lo stato eccitato può essere di singoletto (S) o di tripletto (T). È definito di singoletto se l'elettrone è promosso ad un livello energetico più alto mantenendo il proprio spin; di tripletto se lo spin viene invertito nella promozione.
Il ritorno allo stato fondamentale può avvenire secondo diversi meccanismi:
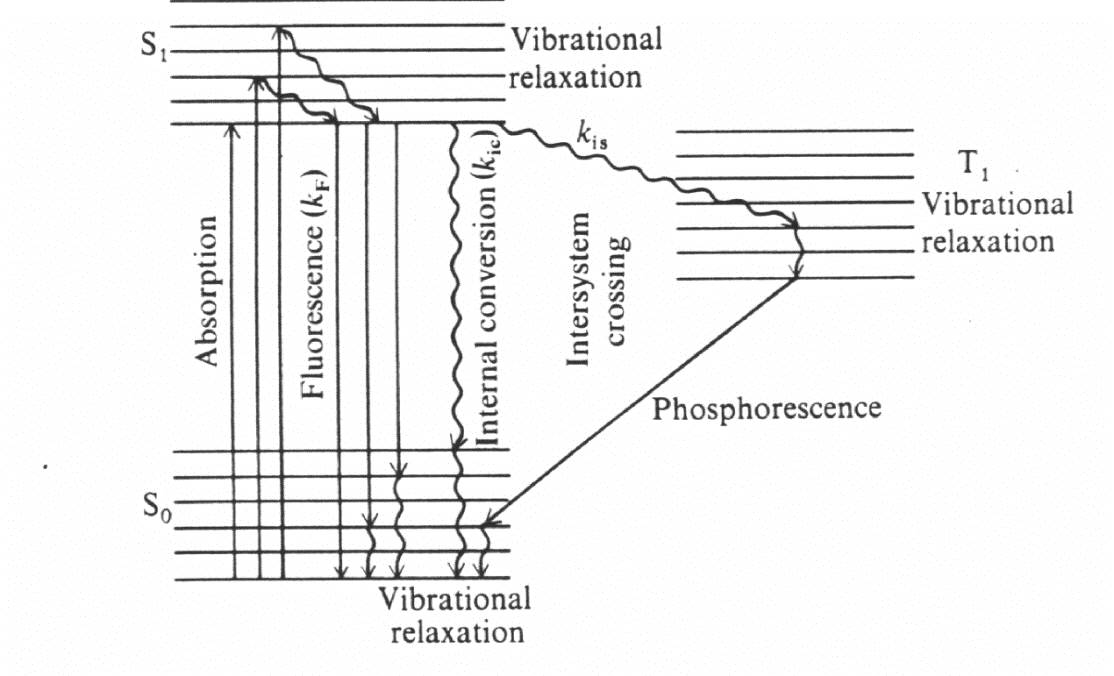 :
Meccanismi di eccitazione e diseccitazione
:
Meccanismi di eccitazione e diseccitazione
I pigmenti sono sostanze in grado di assorbire alcune lunghezze
d'onda della luce visibile (tra 350 e 800 nm). Appaiono quindi colorati
diversamente a seconda delle radiazioni luminose trattenute. Si tratta
di composti chimici che possiedono un esteso sistema di doppi legami coniugati
in cui la delocalizzazione degli orbitali rende la differenza di energia
tra lo stato fondamentale e quello eccitato sufficientemente bassa da far
rientrare la lunghezza d'onda della radiazione associata alla transizione
nell'intervallo della luce visibile.
I pigmenti fotosintetici sono specializzati nell'assorbimento della luce solare e nella sua conversione in energia chimica.
Ne esistono diversi, ciascuno dei quali assorbe in diverse regioni dello spettro: clorofille (tetrapirroli ciclici), carotenoidi e biline (tetrapirroli lineari).
Tale coda non influenza le caratteristiche spettroscopiche ma conferisce alla molecola una consistente idrofobicità.
La presenza di un elevato numero di doppi legami coniugati nel tetrapirrolo è responsabile dell'assorbimento nella regione del visibile.
Le funzioni svolte dalle clorofille sono molteplici:
L'energia luminosa è raccolta come energia di eccitazione
dai pigmenti coordinati da proteine della membrana tilacoidale, dette proteine
antenna.
Questa energia viene convogliata dai complessi antenna ai centri di reazione dove promuove le reazioni fotochimiche primarie.
Il trasferimento è molto efficiente, superiore al 95% (percentuale di fotoni assorbiti trasferiti ai centri di reazione) in condizioni ottimali.
I parametri fondamentali per il trasferimento sono la distanza tra i pigmenti, la loro orientazione e il livello delle transizioni elettroniche (il grado di sovrapposizione dello spettro di emissione del donatore con lo spettro di assorbimento dell'accettore).
Da qui la grande importanza delle proteine antenna cui spetta la responsabilità della creazione e del mantenimento di questi parametri.
Queste infatti non solo mantengono i pigmenti in posizione ottimale per il trasferimento ma, costituendone l'intorno elettronico, ne modulano anche le proprietà spettrali.
Il donatore primario viene nel frattempo ridotto ed è quindi in grado di essere nuovamente ossidato all'arrivo di altra energia di eccitazione dalle antenne. In questo modo l'energia luminosa (assorbita nelle antenne e trasferita al centro di reazione come energia di eccitazione) viene convertita in energia redox e diventa utilizzabile per i processi metabolici cellulari e per un'eventuale nuova conversione ad energia chimica (tramite la fissazione di CO2 a carboidrato).
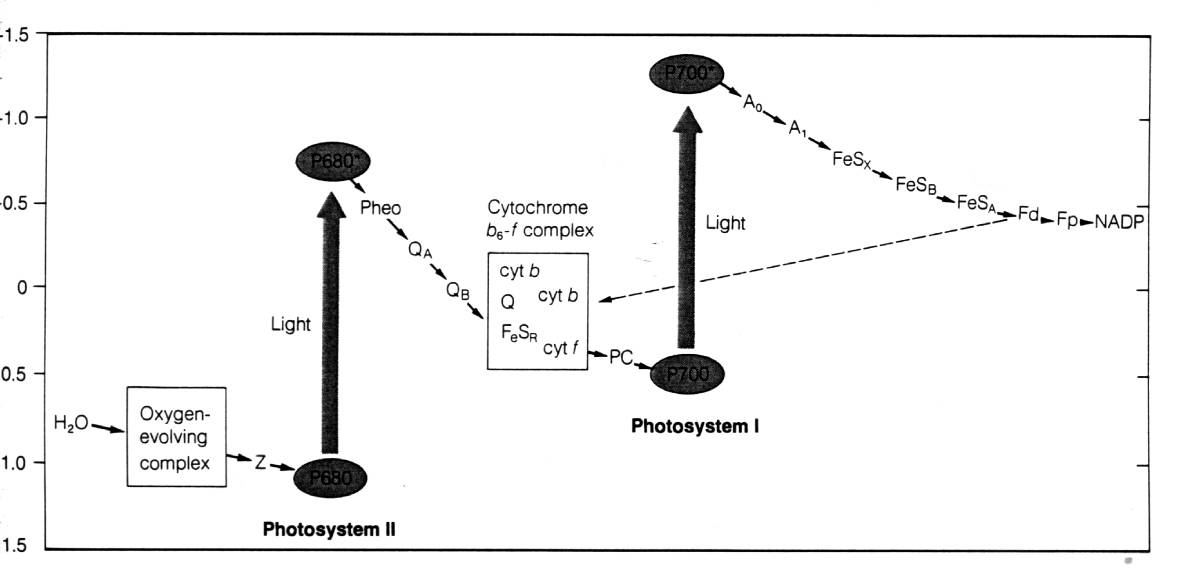
Nella fotosintesi ossigenica, la riduzione del NADP+ e l'ossidazione dell'acqua è attuata da due fotosistemi che agiscono in serie secondo il cosiddetto schema a Z di Hill e Bendall [1960]:
 Figura
-5: Schema a Z di Bendall e Hill. In ordinata il livello di potenziale
redox
Figura
-5: Schema a Z di Bendall e Hill. In ordinata il livello di potenziale
redox
Il primo evento fotochimico nel PS II consiste nella separazione di carica sul P680 (il donatore primario: un dimero di clorofilla denominato P680 dalla lunghezza d'onda a cui assorbe) e quindi nel trasferimento di un elettrone tra questo e l'accettore primario (la feofitina, Pheo in figura) che porta alla formazione di P680+/Pheo-. La ricombinazione di carica è impedita dal veloce trasferimento (200 ps) dell'elettrone dalla feofitina ad una molecola di plastochinone (QA), permanentemente legata al PS II. P680+ possiede un forte potere ossidante e viene riportato allo stato non eccitato estraendo un elettrone dall'acqua e venendo quindi ad essere nuovamente disponibile per la successiva reazione fotochimica.
L'elettrone su QA- è quindi trasferito da una serie di trasportatori (il secondo plastochinone mobile QB ed il complesso del citocromo b6f) alla plastocianina (PC), una proteina diffusibile.
In seguito a foto-ossidazione il PS I (detto P700) catalizza l'ossidazione della plastocianina e la riduzione della ferredoxina (Fd), una piccola proteina stromale che nella sua forma ridotta fornisce gli elettroni per la formazione del NADPH.
Oltre a creare energia redox sotto forma di NADPH, il trasporto elettronico genera un potenziale elettrochimico attraverso la membrana tilacoidale per mezzo di due tipi di reazioni:
Sono pigmenti ampiamente diffusi nel mondo vegetale e si rinvengono, oltre che nel cloroplasto, anche nei cromoplasti, organelli responsabili del colore di fiori e frutti.
Quelli coinvolti nei processi fotosintetici sono detti carotenoidi primari di cui i più abbondanti sono: le xantofille luteina, neoxantina e violaxantina e il b -carotene.
Il loro assorbimento è compreso tra i 350 e 550 nm.
I ruoli ad essi riconosciuti sono [Yamamoto e Bassi 1996]:
I carotenoidi possono impedire la formazione di questa specie chimica poiché il tripletto di clorofilla può trasferire la sua energia di eccitazione ad un carotenoide:
Quest'ultimo ruolo fotoprotettivo in cui i carotenoidi sono coinvolti
permette la dissipazione (quenching) non fotochimica: in condizioni
di luce eccessiva i carotenoidi possono contribuire allo smorzamento dell'energia
in eccesso.
Le proteine dei complessi di raccolta della luce si possono suddividere in quattro classi in base alle caratteristiche delle proteine stesse e del tipo di pigmento fondamentale impiegato:
Come esempi di strutture di proteine antenna procariotiche si riportano
qui due proteine transmembrana e due solubili.
Codice PDB: 1kzu | Figura B-7a
[McDermott et al. 1995]
L'apparato fotosintetico dei batteri purpurei consiste di due tipi di complessi pigmento-proteina: i centri di reazione e i sistemi antenna. Nella maggior parte dei batteri purpurei le membrane fotosintetiche contengono due tipi di sistemi antenna: LH-I (light-harvesting complex I) e LH-II (light-harvesting complex II). Mentre LH-I è fortemente unito ai centri di reazione fotosintetici, LH-II non è ad essi direttamente associato, ma vi trasferisce l'energia di eccitazione tramite LH-I [Zuber et al. 1991].
La struttura di LH-II di Rhodopseudomonas acidophila è stata determinata ad una risoluzione di 2.5 Å.
È una proteina di membrana polimerica composta da monomeri costituiti da due eliche transmembrana. Ogni monomero coordina tre batterioclororofille disposte secondo due differenti orientazioni rispetto alla membrana: con il piano dell'anello macrociclico perpendicolare alla membrana (due) o ad essa parallelo (una). Le prime sono coordinate direttamente da residui di Istidina disposti sulla normale al macrociclo (con una distanza tra l'azoto dell'Istidina e magnesio della clorofilla pari a 2.35 Å), le seconde coordinate sempre da His ma con una molecola d'acqua a ponte (distanza di 2.94 Å tra azoto dell'Istidina e ossigeno dell'acqua, 4.09 Å tra ossigeno dell'acqua e magnesio della clorofilla).
La struttura quaternaria presenta le eliche transmembrana disposte a circolo, coordinando in totale 27 clorofille.
Codice PDB: 1lgh | Figura B-7b
[Koepke et al. 1996]
La struttura dell'LH-II di quest'altro battere purpureo è stata risolta a 2.4 Å.
Presenta un organizzazione circolare (a formare una sorta di barile) di 8 monomeri, ciascuno formato da due eliche transmembrana coordinanti 3 clorofille (2 con macrociclo perpendicolare alla membrana, 1 con macrociclo parallelo ad essa, coordinate allo stesso modo di quanto visto sopra per Rhodopseudomonas acidophila).
Codice PDB: 1ppr | Figura B-7c
[Hofmann et al. 1996]
La PCP (Peridinin-Chlorophyll Protein) è una proteina antenna solubile codificata da questo dinoflagellato.
La sua struttura, determinata ad una risoluzione di 2.0 Å presenta un intreccio di a -eliche coordinanti otto carotenoidi (consentendo a questo organismo di catturare la luce nell'intervallo blu-verde) e due clorofille, con residui Istidina e molecola d'acqua a ponte, come mostrato in figura.
[Tronrud et al. 1986]
La bacteriochlorophyll protein di questo batterio verde è una proteina solubile di peso molecolare 150000 Dalton, composta di tre monomeri identici arrangiati spazialmente secondo un asse ternario di simmetria rotazionale (rotazioni di 120°), la cui struttura è stata determinata ad una risoluzione di 1.9 Å.
Ogni monomero è formato da un largo b-sheet ritorto di 16 filamenti che forma l'esterno (ovvero la parte esposta al solvente) della proteina e racchiude un nucleo centrale di sette batterioclorofille.
Cinque di queste appaiono coordinate da residui Istidina (di cui quattro con l'azoto NE2 ed una - His289 - con l'azoto ND1), una da una molecola d'acqua ed una da un ossigeno dello scheletro polipeptidico (appartenente al residuo Leu234), con una distanza media di 2.1 Å tra magnesio e atomo coordinante.
Nelle pagine seguenti:
a: LH-II di Rhodopseudomonas acidophila
b: LH-II di Rhodospirillum molischianum
c: PCP di Amphidinium carterae
d: bacteriochlorophyll protein di Prosthecochloris
aestuarii
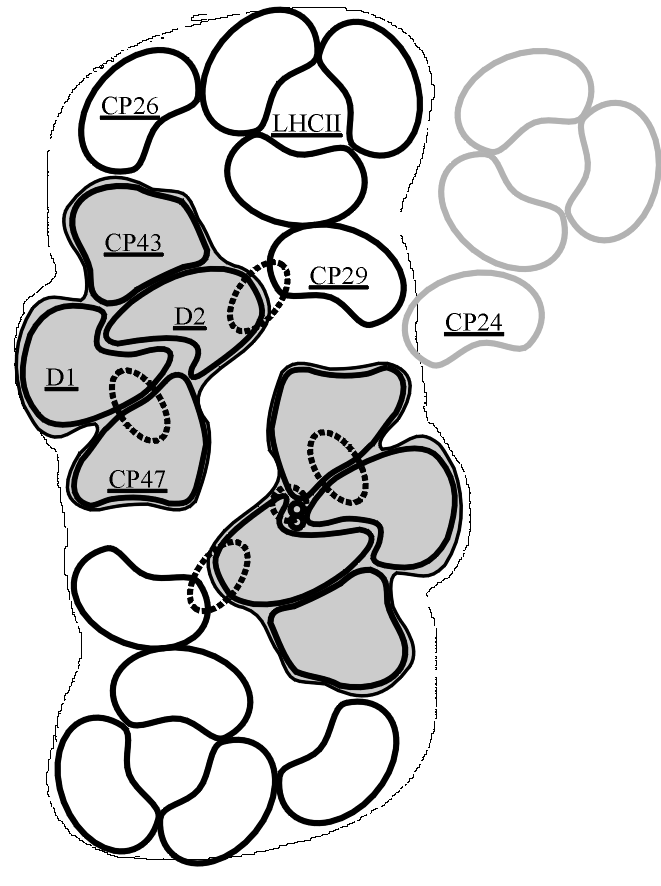
Il PS II contiene due antenne interne, CP43 e CP47 (di 510 e 461
residui aminoacidici) codificate dai geni cloroplastici PsbC e PsbB.
Ciascuna di esse coordina all'incirca venti molecole di clorofilla a e alcune molecole di b -carotene.
Oltre a queste, il PS II si avvale di una serie di complessi antenna più periferici, i cui polipeptidi legano sia clorofilla a che clorofilla b. Per questo motivo sono detti proteine CAB (chlorophyll A/B binding protein).
Sono codificate da geni nucleari, sintetizzate nel citoplasma ed importate nel cloroplasto grazie ad una sequenza leader, il peptide segnale, che viene rimosso al passaggio dell'envelope.
L'analisi delle sequenze aminoacidiche rivela un'elevata omologia, specialmente per due regioni che rappresentano eliche transmembrana, facendo ipotizzare l'esistenza di un gene ancestrale che per duplicazione genica - e per successiva divergenza evolutiva - abbia dato origine all'intera famiglia (Green et al. 1991; cfr. anche § D.V).
La nomenclatura genica identifica Lhca1-4 come i geni responsabili delle proteine antenna legate al PS I e Lhcb1-6 i geni che codificano per il sistema antenna del PS II (cfr. § B.II.7).
I geni Lhcb4, Lhcb5 e Lhcb6 codificano rispettivamente per le cosiddette antenne minori CP29, CP26 e CP24.
I prodotti dei rimanenti Lhcb1, Lhcb2 e Lhcb3 costituiscono il principale complesso antenna, detto LHC II (light harvesting complex del PS II) e sono rispettivamente indicati come LHC II di tipo I, di tipo II e di tipo III.
 Figura
-8: Arrangiamento del PS II proposto da Kilian et al. [1998]
Figura
-8: Arrangiamento del PS II proposto da Kilian et al. [1998]
LHC II è la proteina più abbondante nelle membrane
tilacoidali e lega più della metà delle molecole di clorofilla
in esse presenti.
Grazie a questa sua abbondanza e alla facilità con cui può essere isolato, LHC II è stato il primo complesso antenna ad essere studiato, in campi quali ad esempio l'indirizzamento delle proteine nei cloroplasti e il bilancio dell'energia di eccitazione tra i due fotosistemi.
Il suo ruolo nella ripartizione dell'energia di eccitazione tra i due fotosistemi avviene tramite la migrazione (in seguito a fosforilazione in posizione Thr 3) dai grana (dove normalmente è presente, in posizione periferica rispetto al PS II) alle lamelle stromatiche dove LHC II può connettersi al PS I e aumentarne l'attività rispetto al PS II in condizioni in cui un assorbimento insufficiente di energia luminosa da parte del PS I inibisca il trasporto elettronico [Michel et al. 1990; Allen 1992].
LHC II è presente per lo più in forma trimerica [Peter
e Thornber 1991] e la sua composizione polipeptidica è eterogenea,
essendo sintetizzata da una famiglia multigenica nucleare i cui geni sono
suddivisi nei tre gruppi Lhcb1, Lhcb2, Lhcb3 [Jansson et al. 1992]. È
quindi possibile che l'attività di questo complesso sia controllata
a medio-lungo termine tramite meccanismi di regolazione dell'espressione
genica.
I prodotti dei geni Lhcb1 sono le proteine CAB più abbondanti. In una stessa specie differiscono leggermente l'uno dall'altro [Bassi et al. 1990; Allen e Staehelin 1992]. A seguito della rimozione del peptide segnale tali proteine sono costituite in media da 232 aminoacidi.
Molto omologa al prodotto di Lhcb1 è la proteina prodotta da Lhcb2, meno abbondante e leggermente più corta (mediamente 228 aminoacidi; cfr. § D.V e D.VI per una trattazione approfondita sulla famiglia multigenica, sull'omologia nelle sequenze e sulle caratteristiche di queste).
Si ritiene che i trimeri di LHC II si formino per associazione casuale di proteine di tipo I e II. La trimerizzazione richiede la presenza di un particolare fosfolipide (il fosfatidilglicerolo) contenente acido trans-esadecenoico [Nussberger et al. 1993].
Il dominio idrofilico N-terminale delle due classi di polipeptidi sembra essere coinvolto nella formazione o nella stabilizzazione del trimero poiché esperimenti di delezione hanno mostrato l'incapacità di trimerizzazione in sua assenza. In particolare la sequenza "WYxxxR" (x: aminoacido qualsiasi) è stata identificata come "motivo di trimerizzazione" e potrebbe rappresentare un sito per il legame del fosfolipide [Hobe et al. 1995].
I prodotti dei geni Lhcb3 sono molto meno abbondanti e più corti degli altri due tipi (circa 223 aminoacidi) e da questi presentano una certa divergenza evolutiva.
L'analisi HPLC (high performance liquid chromatography) di complessi
purificati ha messo in evidenza la presenza di luteina come principale
carotenoide, seguita da neoxantina e violaxantina, oltre che da clorofilla
di tipo a e b (cfr. § B.II.4).
Sulla base di studi biochimici, che hanno stabilito un rapporto fra clorofille a/b di 1.4 e una stechiometria di 12 clorofille per polipeptide, si può stimare che ciascun monomero coordini 7 molecole di clorofilla a, 5 di clorofilla b e tre carotenoidi [Bassi et al. 1993].
LHC II è uno dei pochi complessi integrali di membrana - e la prima proteina antenna di pianta superiore - di cui sia nota la struttura, ottenuta mediante cristallografia elettronica su cristalli bidimensionali ad una risoluzione di 3.4 Å.
È un trimero a simmetria rotazionale con asse ternario (C3) ed il cristallo appartiene al gruppo spaziale P 321.
Ogni monomero è costituito - come previsto dall'analisi di sequenza e dagli algoritmi di predizione (vedi § B.V.3.2) - da tre a -eliche transmembrana (denominate B, C e A) e da una quarta corta elica anfipatica in posizione C-terminale (elica D).
La prima e la terza elica transmembrana (rispettivamente B e A) partendo dall'N-terminale formano all'interno della membrana una struttura a forma di X con un asse di simmetria rotazionale binaria (C2); sono altamente omologhe e costituite rispettivamente da 34 e 29 aminoacidi (prolungandosi rispettivamente per 51 e 43 Å, 9,5 e 8 giri di elica).
Ciascuna di esse forma un angolo di 32° con la normale al piano della membrana.
La terza elica, C, è invece praticamente ortogonale (81°) a tale piano ed è costituita da 20 aminoacidi per un'estensione di 31 Å e 5,5 giri di elica.
L'elica D, di soli 10 aminoacidi, è parallela al piano della membrana e si affaccia sul lato lumenale della membrana tilacoidale.
I loop di connessione tra le eliche non appaiono nella struttura cristallografica a causa del fatto che i cristalli sono bidimensionali (e quindi con risoluzioni diverse per le tre dimensioni) o per una debole densità elettronica in queste regioni. I primi 54 residui del polipeptide ed il loop che connette le eliche C ed A (Ala144-Asp169) si trovano nel lato stromatico mentre il loop tra le eliche B e C (Val90-Gln122) e la porzione C-terminale (Asp215-Lys232) sono sul lato lumenale.
La struttura evidenzia la presenza di 12 clorofille e due carotenoidi, ipotizzati essere due luteine (sulla base della stechiometria 2:1 delle luteine per monomero). La risoluzione della struttura cristallografica non è sufficiente a determinare se una molecola di clorofilla sia di tipo a o b, né a discriminare tra le diverse xantofille.
La posizione di questi pigmenti è chiara mentre non è visibile il terzo carotenoide che dai dati biochimici questa proteina dovrebbe coordinare (con una stechiometria per monomero di circa 1 molecola di neoxantina e 0.1-0.2 di violaxantina).
Forse quest'ultimo si trova in posizione più disordinata, meno stabile e quindi meno visibile ai raggi X, oppure potrebbe non essere presente in tutti i monomeri, vista l'eterogeneità nella composizione polipeptidica. Un'altra ipotesi è che la procedura di cristallizzazione comporti la perdita di uno o più carotenoidi.
I due carotenoidi presenti nella struttura cristallografica sono disposti al centro del complesso parallelamente alle eliche A e B, in conformazione a croce rispetto al piano della membrana (con un angolo di 50° con la sua normale). Le teste dei carotenoidi sono praticamente equidistanti da entrambe le superfici di membrana e ad una distanza dai loop colleganti le eliche tale da permettere un'interazione ponte idrogeno. Un carotenoide è probabilmente attaccato alla Glutammina 197 sul lato lumenale e ad un residuo tra Ser160 e Leu164 sul lato stromatico. L'altro può essere legato a siti tra Asp47 e Ala49 sul lato stromatico e tra Trp97 e Ala100 sul lato lumenale. I siti leganti le xantofille sarebbero quindi sulle porzioni conservate di estensione simmetrica alle eliche B ed A, con sequenze consenso probabilmente derivate dalla sequenza WFDPL [Pichersky e Jansson 1996] (cfr. § D.V.1 e D.VI).
I due carotenoidi fornirebbero un forte collegamento tra i loop alle due superfici, probabilmente necessario data la struttura relativamente "aperta" della proteina, dedicata soprattutto alla coordinazione dei pigmenti limitando quindi ad una sola le strette interazioni elica-elica riscontrate nelle strutture procariotiche risolte (cfr. § B.II.5.1). Tale ruolo strutturale centrale delle xantofille spiegherebbe la loro necessità nella ricostituzione in vitro del complesso.
Le clorofille si trovano approssimativamente su due livelli paralleli alla superficie della membrana (sopra e sotto il piano mediano di essa), con l'anello porfirinico quasi perpendicolare alla stessa. Le distanze tra clorofille di uno stesso "piano" variano tra i 9 e i 15 Å, tra 13 e 14 Å quelle di piani diversi.
La struttura cristallografica mette in evidenza alcuni possibili residui aminoacidici coordinanti le clorofille. Al contrario delle strutture precedentemente studiate (i sistemi antenna procariotici), dove le clorofille sono coordinate solo da residui Istidina (con o senza una molecola d'acqua a ponte; cfr. § B.II.5.1), in LHC II le clorofille appaiono coordinate nell'atomo di Magnesio centrale dalle catene laterali di aminoacidi polari. In particolare sono stati identificati i seguenti ligandi:
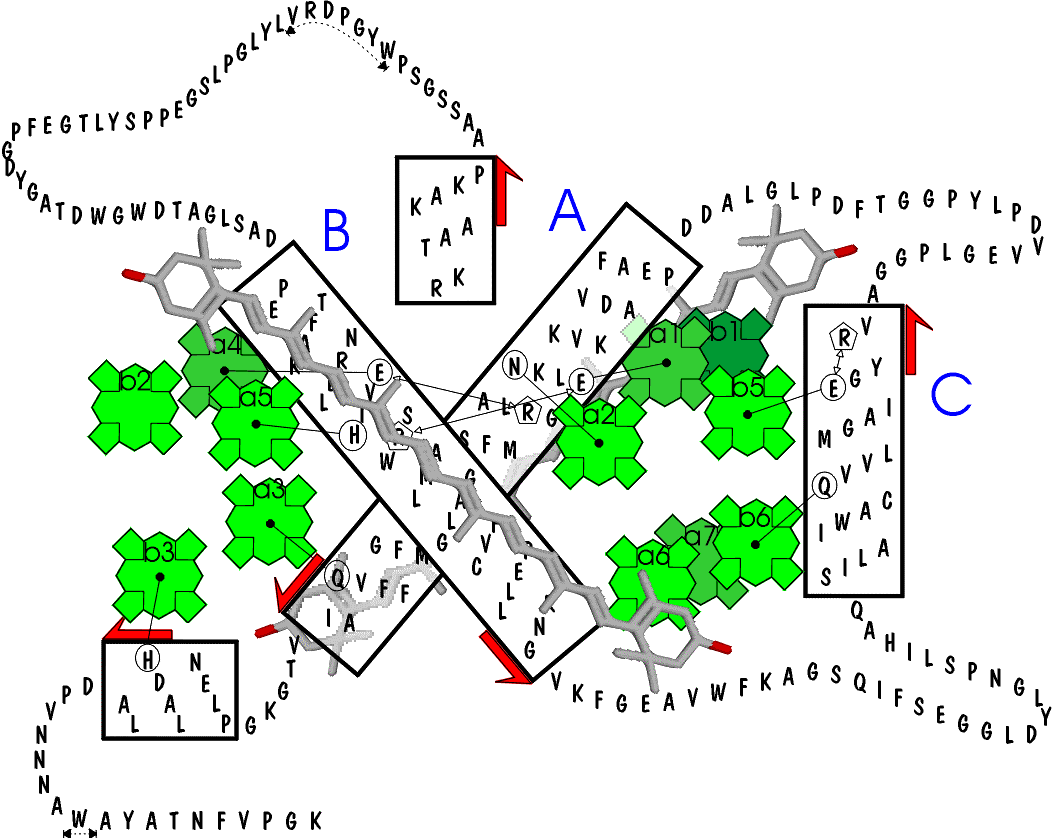 Per le rimanenti clorofille
(b1, b2 e a7) non è chiara la coordinazione.
Per le rimanenti clorofille
(b1, b2 e a7) non è chiara la coordinazione.
Figura -9: Rappresentazione schematica della disposizione e coordinazione dei pigmenti in LHC II
La nomenclatura dei siti è quella usata da Kühlbrandt [Kühlbrandt et al. 1994] che ha distinto i siti a e b come quelli che potrebbero probabilmente ospitare clorofilla di tipo a e b poiché si ritiene che siano le clorofille a a trasferire l'energia di eccitazione ai carotenoidi e quindi siano quelle ad essi più vicine. Questo in base al fatto che il trasferimento di energia da clorofilla b ad a è molto più veloce (sull'ordine dei picosecondi) del tempo di vita dei tripletti di clorofilla (vari nanosecondi); lo smorzamento del tripletto realizzato dai carotenoidi (cfr. § B.II.4.2) è quindi principalmente richiesto per la clorofilla a.
In relazione alla clorofilla a6: questa sembra essere coordinata dal carbonile peptidico della Glicina 78 che non può formare ponte idrogeno - come normalmente avviene nelle a -eliche - con il residuo presente nel giro successivo perché questo è una Prolina (Pro 82). Si noti inoltre che la Glicina dispone di una più elevata libertà conformazionale rispetto a tutti gli altri residui. La distanza tra il Magnesio della clorofilla a6 e il carbonile sembra essere troppo elevata (circa 4.6 Å) per una coordinazione diretta, suggerendo il coinvolgimento di una molecola d'acqua (come in alcune strutture procariotiche, cfr. § B.II.5.1).
Nella pagina seguente:
Questi complessi antenna, localizzati tra il core complex e le antenne
più periferiche, sono presenti nel PS II come monomeri, con una
stechiometria di 1:1 rispetto al centro di reazione (vedi Figura B-8).
Codificata dal gene Lhcb4, è la più lunga delle proteine
CAB (257 aminoacidi) a causa di un inserzione di circa 40 residui aminoacidici
immediatamente a monte dell'elica B rispetto alle altre proteine della
famiglia genica.
Coordina 6 molecole di clorofilla a, due di clorofilla b, una luteina e quantità substechiometriche di violaxantina e neoxantina per un totale di due carotenoidi [Giuffra et al. 1996].
La porzione N-terminale, esposta allo stroma, può essere fosforilata in condizioni di eccessiva illuminazione in posizione 83 [Testi et al. 1996] da una chinasi diversa da quella attiva nella fosforilazione di LHC II [Bergantino et al. 1995]. La fosforilazione induce variazioni conformazionali e riarrangiamento dei pigmenti che potrebbero favorire meccanismi di dissipazione termica dell'energia [Croce et al. 1996].
L'apoproteina codificata dal gene Lhcb5 è composta di 247
residui aminoacidici e coordina 6 clorofille a, 3 di tipo b, una luteina
e quantità substechiometriche di violaxantina e neoxantina per un
totale di due carotenoidi [Ros et al. 1998].
CP 26 sembrerebbe coinvolto nel processo di dissipazione termica dell'energia, tramite la protonazione di un Glutammico che causerebbe una riorganizzazione dei pigmenti nella porzione proteica prossimale al lumen, meccanismo comune a CP 29. [Walters et al. 1994].
Le proteine CAB (chlorophyll a/b binding) del fotosistema I e II sono rispettivamente codificate da geni indicati come Lhca e Lhcb (la maiuscola iniziale si riferisce a geni codificati nel nucleo).
I quattro distinti tipi di proteine CAB identificati nel complesso antenna LHC I sono codificati da geni identificati con le sigle Lhca1 Lhca2 Lhca3 e Lhca4.
I tre tipi di proteine del complesso antenna maggiore LHC II (dette LHC II di tipo I, II e III) sono codificate dai geni Lhcb1 Lhcb2 e Lhcb3.
I geni Lhcb4, Lhcb5 e Lhcb6 codificano per le antenne minori del fotosistema II: CP 29, CP 26 e CP 24 rispettivamente. Questa nomenclatura proteica si riferisce al peso apparente (in kD) valutato attraverso la mobilità relativa (dalla più lenta alla più veloce) delle proteine codificate in SDS-PAGE.
L'evoluzione proteica è il motore per l'evoluzione delle
specie viventi che si sono tutte sviluppate a partire da una singola o
da un numero molto limitato di specie ancestrali.
L'evoluzione delle proteine si attua per mezzo di cambiamenti di singoli residui, inserzioni e delezioni di parecchi residui, duplicazione genica, fusione genica. Nel corso del tempo queste modificazioni si accumulano fino al punto di perdere ogni similarità tra la sequenza iniziale e quella risultante.
La tendenza alla sostituzione di un residuo ad una data posizione aminoacidica può variare considerevolmente.
Estesi cambiamenti possono risultare dalla duplicazione genica, che può portare al raddoppiamento in lunghezza della catena polipeptidica. Inoltre si conoscono casi di fusione di diversi geni strutturali, in cui uno o più geni sono stati traslocati in una diversa posizione nel genoma.
Gli studi sull'evoluzione di proteine omologhe in organismi diversi permettono la ricerca della genealogia delle specie. In generale, infatti, le sequenze aminoacidiche e i motivi caratteristici delle strutture terziarie sono conservati al punto che proteine di specie anche molto distanti si assomigliano le une alle altre. Ne consegue che tali analisi filogenetiche possono rappresentare un metodo di tassonomia [Schulz e Schirmer 1979].
La diversificazione funzionale di proteine omologhe - detta anche differenziazione proteica - mostra che la diversità biologica è limitata: le proteine possono essere classificate su linee generali [Dayhoff 1976].
La base dell'evoluzione proteica è rappresentata dalle mutazioni sul DNA. A livello proteico la velocità di fissazione di una mutazione è generalmente espresso con una misura della percentuale di mutazioni puntiformi accettate in 108 anni. Sono le cosiddette unità PAM (Percentage of Accepted point Mutation), usate comunemente per misurare la distanza evolutiva.
Molti sono i fattori che influenzano l'importanza biologica dei singoli residui all'interno della sequenza proteica e quindi il maggiore o minore contributo dato da questi al processo di evoluzione. I vari aminoacidi possono essere indispensabili per mantenere la funzione della proteina (ad esempio nel sito attivo di un enzima) o a regolarne l'attività (come ad esempio gli aminoacidi costituenti un sito allosterico di controllo di un enzima) o a caratterizzarne la struttura (specifici ripiegamenti nella struttura derivanti dall'ingombro sterico di un particolare aminoacido) o le proprietà globali (come l'idrofobicità o la carica nelle diverse parti della proteina).
Come detto sopra è stato osservato come gli aminoacidi presenti alla superficie delle proteine siano più esposti a cambiamento. Eccezioni a questo sono i residui di superficie che siano coinvolti nell'attività e nelle interazioni della proteina.
Gli effetti di un cambiamento aminoacidico all'interno delle proteine sono spesso compensati da altri cambiamenti (cfr. § B.VII). Un esempio molto elaborato di compensazione interna si osserva in due diverse serine-proteasi imparentate: il nucleo interno composto da Trp29, Ser45, Val53, Val200, Leu209, Val210 e Ile212 nella chimotripsina diventa Ser29, Thr45, Met53, His200, Val209, His210, Val212 nell'elastasi, senza modificazioni a livello di scheletro polipeptidico delle catene proteiche [Hartley 1970].
La conservazione della struttura o della funzione restringe i cambiamenti permessi in una data posizione aminoacidica. Alcune modificazioni sono meno rilevanti e possono essere sopportate più facilmente (e quindi trasmesse, fissate) dalla proteina. Queste sono definite sostituzioni conservative, ovvero modificazioni tra residui simili. A questo proposito sono rilevanti caratteristiche quali la dimensione, la forma, la flessibilità, la carica di una catena laterale, nonché la sua capacità di formare ponti idrogeno o la sua idrofobicità.
Le probabilità di mutazione per i diversi tipi aminoacidici possono essere calcolate e tabulate. Le tabelle di probabilità di mutazione riportano la probabilità per ogni tipo aminoacidico di mutare in ogni altro tipo. I termini sulla diagonale indicano la probabilità che il residuo non vada incontro a mutazioni, e resti invariato.
Ad esempio il residuo Serina ha un alta probabilità di mutazione. Questo aminoacido si trova normalmente alla superficie proteica. Il residuo Triptofano ha la minore probabilità di mutazione. Questo è ragionevole poiché esso è generalmente un residuo interno e non può essere sostituito da una catena laterale di uguale ingombro sterico.
Queste tabelle sono alla base per la costruzione delle matrici di sostituzione (cfr. § B.V.2.1).
In realtà questo è un discorso generale che può essere meno applicabile ad alcuni sistemi. Per esempio gli aminoacidi idrofobici che compongono eliche transmembrana, che non abbiano funzioni particolari, mutano ma mantengono carattere idrofobico.
Queste due parole ricorrono sovente nel corso di questa tesi. Segue
quindi la loro definizione e una spiegazione sul vario uso che di esse
viene fatto.
Omologia: Corrispondenza fra strutture in organismi derivanti da una forma ancestrale comune, a prescindere dalla funzione.
Analogia: Corrispondenza nella funzione anche se con origine molto diversa.
Si parla dunque di modellistica per omologia perché l'alta omologia insita nel concetto di famiglia proteica è un prerequisito (cfr. § B.VI). Sia orizzontalmente, ovvero per diverse proteine dello stesso organismo, che verticalmente, ovvero per la stessa proteina in diverse specie.
Per quanto riguarda la similarità aminoacidica, questa verrà qui distinta in Identità e in Analogia, per evitare l'ambiguità generalmente presente in letteratura dove spesso la definizione "sequenze simili" viene applicata sia a sequenze con un alto grado di aminoacidi allineati identici che a sequenze i cui aminoacidi, pur non essendo uguali, siano simili come caratteristiche fisico-chimiche (cfr. § B.III).
Si considera identità - come è logico aspettarsi - l'uguale corrispondenza di un residuo ad una certa posizione nelle due (o più) sequenze analizzate. Viene invece chiamata analogia la presenza nelle sequenze di residui che svolgano la stessa funzione o che abbiano carattere simile (come l'idrofobicità o le dimensioni steriche) e che quindi contribuiscano alla similarità tra le sequenze, ovvero alla loro probabile omologia.
E.g.:
| EARSW EARSW
EARSW DVKTY Identità Analogia |
La bioinformatica è una branca della disciplina nota come
scienza computazionale che utilizza le potenzialità di calcolo e
visualizzazione grafica dei computer per studiare problemi in biologia,
chimica, matematica, fisica ed altri campi.
La ricerca nei campi della biologia e genetica molecolare sta generando un incredibile quantità di dati che non possono essere analizzati manualmente. Ad esempio GenBank, banca dati genetica, contiene 3.27 milioni di sequenze da più di 35000 specie (al Marzo 1999) e raddoppia in meno di un anno.
Le tecnologie informatiche sono utilizzate per raccogliere, organizzare ed analizzare tali dati.
L'analisi delle sequenze genetiche o proteiche, l'analisi delle strutture tridimensionali di proteine, le simulazioni di meccanica e dinamica molecolare, la ricerca conformazionale, la modellistica per omologia sono alcune delle possibilità offerte dalla bioinformatica.
Grazie allo sviluppo di Internet ed al suo sfruttamento da parte
del mondo scientifico sono presenti in essa, e liberamente consultabili,
vari database (basi di dati): collezioni ordinate di informazioni,
interrogabili tramite varie sintassi di ricerca. Importantissimi per le
scienze biomediche sono i database che contengono sequenze, strutture proteiche,
interi genomi, sequenze segnale.
Accesso ai database: in linea di principio vi sono due distinti modi per accedere ai database pubblici. Uno è di visualizzare i file tramite il protocollo WWW, l'altro è di copiare i file sulla propria macchina locale (solitamente tramite protocollo FTP).
Un programma importante disponibile su WWW è l'SRS (Sequence Retrieval System [Etzold et al. 1996]), un sistema di recupero informazioni che facilita la ricerca in vari database di sequenza adattando la richiesta dell'utente ai diversi formati di interrogazione di questi.
Database nucleotidici: i due principali sono mantenuti all'EMBL-EBI in Inghilterra (EMBL nucleotide db [Shomer et al. 1996]) e all'NCBI negli Stati Uniti (GenBank [Benson et al. 1996]). Altri database sono specifici per alcune specie studiate in progetti genoma.
Database proteici: i maggiori per le sequenze sono SWISS-PROT (Basel in Svizzera, [Bairoch e Apweiler 1996]) - che contiene anche il TREMBL (Translated EMBL: la traduzione del DNA codificante del database EMBL in sequenze aminoacidiche) - e il PIR [George et al. 1996].
Informazioni sulla struttura 3D sono contenute nel PDB [Bernstein et al. 1997] e nei database da esso derivati (e.g. HSSP - allineamenti delle sequenze delle proteine di SwissProt le proteine contenute nel PDB - e FSSP - allineamenti strutturali delle proteine contenute nel PDB).
Questi database riportano sempre annotazioni nelle sequenze o strutture che essi contengono, permettendo di accedere a multiple informazioni quali la data di immissione, l'autore, metodi, nomenclatura.
Una possibilità interessante fornita dalla presenza di queste collezioni di dati è quella di confrontare una sequenza (o una struttura) con tutte quelle a lei simili, già catalogate.
A livello proteico la pressione evolutiva selettiva proviene dalla
necessità di mantenere la funzione e questo a sua volta implica
un mantenimento della specifica struttura 3D. Questa è la base per
l'allineamento delle sequenze proteiche, ovvero il rilevamento ottimale
delle posizioni equivalenti in stringhe di aminoacidi.
L'allineamento porta alla luce informazioni sulle relazioni strutturali e funzionali tra i residui di differenti proteine.
L'obiettivo è di trovare la miglior corrispondenza tra due stringhe di lettere (siano esse i codici per le basi nucleotidiche o per gli aminoacidi).
Vi sono algoritmi di ricerca veloce (utili per una scansione completa dei database esistenti al fine di trovare sequenze simili alla sequenza sotto esame) e altri - più lenti - per realizzare allineamenti accurati.
L'arte dell'allineamento consiste nell'allineare segmenti correlati ed evitare di allineare segmenti di sequenze senza nessuna relazione [Deperieux e Feytmans 1992; Eddy 1995; Henikoff e Henikoff 1994; Krogh, et al. 1994; Lawrence, et al. 1993; Livingstone e Barton 1993; Russell e Barton 1992; Sander e Schneider 1991; Thompson, et al. 1994]
Il maggiore problema nel comparare diverse procedure di allineamento è la mancanza di criteri riconosciuti per misurare la qualità di un allineamento.
Spesso l'allineamento migliore dipende dall'obiettivo che ci si prefigge. Se si cercano omologie strutturali saranno preferiti gli allineamenti globali, quelli cioè che identificano maggiori similarità tra le sequenze. Per la ricerca di omologie funzionali saranno spesso preferiti gli allineamenti locali che puntino al sito attivo responsabile della funzione.
Inoltre vi è l'allineamento migliore definito non biologicamente ma matematicamente, ovvero quello che massimizza una data funzione bersaglio, ad esempio trovando l'allineamento con il massimo numero di coppie di residui identici.
Un elemento rilevante è la trattazione dei gap ovvero dei residui inseriti o rimossi per ottimizzare la funzione bersaglio.
Tutto viene valutato in base ad un punteggio ("score"). L'algoritmo di allineamento cercherà di trovare blocchi di omologia e di estenderli il più possibile. L'estensione delle zone simili viene premiata con un punteggio positivo per il particolare allineamento. La creazione di un gap e la sua estensione presentano punteggio negativo (tipicamente la penalità per l'estensione è 5-10 volte inferiore al costo dell'introduzione).
Le penalità possono essere configurate dagli utenti in modo da calibrare l'algoritmo e indirizzarlo. Ad esempio abbassando la penalità al "gap open" si favorisce la ricerca di omologia diffusa, permettendo al programma di creare vari gap. Il rischio implicito è quello di superare il limite tra ciò che ha significato biologico e ciò che ne è sprovvisto.
A modificazione del punteggio (e quindi ad ulteriore intralcio nei tentativi di confronto tra le diverse procedure) interviene la differenza di valutazione tra residui identici e residui analoghi.
Alcune sostituzioni aminoacidiche (e.g. I ®
L) sono praticamente neutrali per quanto riguarda il mantenimento della
struttura o della funzione.
Questo porta a basare l'allineamento su matrici di sostituzione che rappresentino le proprietà fisico chimiche e la differente probabilità statistica dei residui aminoacidici (e.g. Ser ha un alta mutabilità al contrario di Trp). Molte matrici sono disponibili [Feng, et al. 1985; McLachlan 1972; Dayhoff 1978; Bowie, et al. 1991; Gonnet, et al. 1992; Gribskov, et al. 1990; Henikoff e Henikoff 1994; Overington, et al. 1990; Risler, et al. 1988; Taylor 1986; Thompson, et al. 1994] e decidere quale usare può essere un dilemma. J. e S. Henikoff [Henikoff e Henikoff 1993] hanno sistematicamente confrontato le prestazioni di varie matrici e sono giunti alla conclusione che nessuna matrice è a priori la migliore per allineare una sequenza data, anche se la "BLOSUM62" è risultata mediamente la migliore nei loro test. È utile provare l'allineamento con varie matrici per confrontare il loro comportamento nel caso in esame.
Come esempio si riporta qui la BLOSUM62 [Henikoff e Henikoff 1992] indicante in forma di matrice quadrata le variazioni al punteggio da applicare in un allineamento in relazione al confronto tra aminoacidi su due sequenze (segnate in grassetto le posizioni relative alle ricorrenze S® S=4 e W® W=11 che mostrano la maggior importanza data alla conservazione del Triptofano - e la minore data alla Serina - a causa della frequenza di mutazione di questi due residui, cfr. § B.III):
scores
{
title "BLOSUM 62",
seq-type amino,
symbol-set "ARNDCQEGHILKMFPSTWYVBZX",
score-table
{
{ 4,-1,-2,-2, 0,-1,-1, 0,-2,-1,-1,-1,-1,-2,-1, 1, 0,-3,-2, 0,-2,-1, 0 },
{-1, 5, 0,-2,-3, 1, 0,-2, 0,-3,-2, 2,-1,-3,-2,-1,-1,-3,-2,-3,-1, 0,-1 },
{-2, 0, 6, 1,-3, 0, 0, 0, 1,-3,-3, 0,-2,-3,-2, 1, 0,-4,-2,-3, 3, 0,-1 },
{-2,-2, 1, 6,-3, 0, 2,-1,-1,-3,-4,-1,-3,-3,-1, 0,-1,-4,-3,-3, 4, 1,-1 },
{ 0,-3,-3,-3, 9,-3,-4,-3,-3,-1,-1,-3,-1,-2,-3,-1,-1,-2,-2,-1,-3,-3,-2 },
{-1, 1, 0, 0,-3, 5, 2,-2, 0,-3,-2, 1, 0,-3,-1, 0,-1,-2,-1,-2, 0, 3,-1 },
{-1, 0, 0, 2,-4, 2, 5,-2, 0,-3,-3, 1,-2,-3,-1, 0,-1,-3,-2,-2, 1, 4,-1 },
{ 0,-2, 0,-1,-3,-2,-2, 6,-2,-4,-4,-2,-3,-3,-2, 0,-2,-2,-3,-3,-1,-2,-1 },
{-2, 0, 1,-1,-3, 0, 0,-2, 8,-3,-3,-1,-2,-1,-2,-1,-2,-2, 2,-3, 0, 0,-1 },
{-1,-3,-3,-3,-1,-3,-3,-4,-3, 4, 2,-3, 1, 0,-3,-2,-1,-3,-1, 3,-3,-3,-1 },
{-1,-2,-3,-4,-1,-2,-3,-4,-3, 2, 4,-2, 2, 0,-3,-2,-1,-2,-1, 1,-4,-3,-1 },
{-1, 2, 0,-1,-3, 1, 1,-2,-1,-3,-2, 5,-1,-3,-1, 0,-1,-3,-2,-2, 0, 1,-1 },
{-1,-1,-2,-3,-1, 0,-2,-3,-2, 1, 2,-1, 5, 0,-2,-1,-1,-1,-1, 1,-3,-1,-1 },
{-2,-3,-3,-3,-2,-3,-3,-3,-1, 0, 0,-3, 0, 6,-4,-2,-2, 1, 3,-1,-3,-3,-1 },
{-1,-2,-2,-1,-3,-1,-1,-2,-2,-3,-3,-1,-2,-4, 7,-1,-1,-4,-3,-2,-2,-1,-2 },
{ 1,-1, 1, 0,-1, 0, 0, 0,-1,-2,-2, 0,-1,-2,-1, 4, 1,-3,-2,-2, 0, 0, 0 },
{ 0,-1, 0,-1,-1,-1,-1,-2,-2,-1,-1,-1,-1,-2,-1, 1, 5,-2,-2, 0,-1,-1, 0 },
{-3,-3,-4,-4,-2,-2,-3,-2,-2,-3,-2,-3,-1, 1,-4,-3,-2,11, 2,-3,-4,-3,-2 },
{-2,-2,-2,-3,-2,-1,-2,-3, 2,-1,-1,-2,-1, 3,-3,-2,-2, 2, 7,-1,-3,-2,-1 },
{ 0,-3,-3,-3,-1,-2,-2,-3,-3, 3, 1,-2, 1,-1,-2,-2, 0,-3,-1, 4,-3,-2,-1 },
{-2,-1, 3, 4,-3, 0, 1,-1, 0,-3,-4, 0,-3,-3,-2, 0,-1,-4,-3,-3, 4, 1,-1 },
{-1, 0, 0, 1,-3, 3, 4,-2, 0,-3,-3, 1,-1,-3,-1, 0,-1,-3,-2,-2, 1, 4,-1 },
{ 0,-1,-1,-1,-2,-1,-1,-1,-1,-1,-1,-1,-1,-1,-2, 0, 0,-2,-1,-1,-1,-1,-1 }
}
}
Questo garantisce un allineamento matematicamente ottimo, relativo ad una matrice di sostituzione e alle penalità scelte per i gap.
La generalizzazione della programmazione dinamica agli allineamenti multipli è limitata ad un basso numero di sequenze piuttosto corte [Lipman et al. 1989].
Per più di circa 8 proteine di media lunghezza il problema oltrepassa le attuali capacità di calcolo.
Correntemente l'approccio più diffuso è lo sfruttamento del fatto che le sequenze omologhe sono evolutivamente correlate.
Si può quindi costruire un allineamento multiplo progressivamente con una serie di allineamenti a coppie, seguendo l'ordine dei rami di un albero filogenetico [Feng e Doolittle 1987]. Prima vengono allineate le sequenze più vicine, aggiungendo gradualmente le più distanti.
Questo metodo è sufficientemente veloce da permettere virtualmente allineamenti di qualunque estensione.
La predizione di alcuni importanti parametri della struttura 3D
(ad esempio la struttura secondaria, l'accessibilità al solvente,
le eliche transmembrana) a partire dalle informazioni di sequenza è
un compito molto più semplice rispetto alla modellistica per omologia.
Questo è reso evidente dall'alto numero di servizi online nati dopo che il primo di essi (PredictProtein) fu offerto nel 1992. Pochi però sono sufficientemente testati ed affidabili.
Il concetto alla base della maggior parte dei metodi per la predizione
della struttura secondaria è la preferenza di segmenti di residui
consecutivi per certi stati di struttura secondaria [Kabsch e Sander 1984].
Il problema di predizione diventa un problema di classificazione di domini strutturali. L'obiettivo è di predire se il residuo al centro di un segmento - tipicamente della lunghezza di 13-21 residui - appartenga ad un'elica, un foglietto beta o ad una struttura secondaria non regolare [Barton 1995; Garnier, et al. 1996; Rost e Sander 1993; Rost e Sander 1996; Rost et al. 1993].
Basando le predizioni su sequenze singole l'accuratezza di predizione è limitata a circa il 60% (percentuale di residui correttamente predetti). Usando come input un allineamento multiplo si supera il 72% di accuratezza.
Tali predizioni possono essere usate, ad esempio, per assegnare la classe strutturale di una proteina (tutte eliche, tutti strand beta...).
Poiché le regioni intra- ed extra-citoplasmiche hanno diversa composizione aminoacidica [Nakashima e Nishikawa 1992; von Heijne 1992], è possibile predire l'orientazione delle eliche transmembrana rispetto alla membrana (N-terminale che punti verso l'interno o l'esterno).
Questo tipo di predizioni costituiscono un interessante strumento per l'analisi di interi genomi (alcune ore di calcolo sono sufficienti per la scansione dell'intero genoma di Haemophilus influenzae, ad esempio) per classificare le proteine che contengano le eliche transmembrana e quelle che ne siano prive. La quantità di falsi positivi (proteine senza alcuna elica transmembrana osservata che siano predette contenerne) è sotto il 2% mentre i falsi negativi (proteine contenenti eliche transmembrana che non siano predette averle) ammontano a circa il 3% [Rost et al. 1996].
La conoscenza della struttura tridimensionale di una proteina è di grande aiuto nel pianificare esperimenti mirati alla comprensione della sua funzione o nella progettazione di composti mirati ad un'interazione specifica con la proteina (comunemente detto "drug design").
Purtroppo la disponibilità della struttura delle proteine è spesso mancante a causa delle difficoltà nell'ottenere sufficiente proteina pura, cristalli capaci di diffrangere o sistemi di sovraespressione per produrre proteine (eventualmente marcate in 15N e 13C) per studi NMR.
Questo è particolarmente rilevante per le proteine di membrana.
Ne consegue che il numero di proteine risolte aumenta molto lentamente in confronto con la mole di nuove sequenze e nessuna informazione strutturale è disponibile per la maggioranza delle sequenze proteiche contenute nei numerosi database.
Da qui all'importanza assunta dai metodi predittivi il passo è breve.
Proteine provenienti da differenti fonti e a volte anche con diversa funzione biologica possono avere sequenze simili ed è generalmente accettato che un'alta similarità di sequenza corrisponda ad una ben distinta similarità di struttura.
La deviazione quadratica media (rmsd) delle coordinate dei carboni alfa per core (parti più interne) proteici che condividano il 50% di identità di sequenza è prevista essere circa 1Å.
Questo fatto è alla base dello sviluppo del modelling (modellistica, costruzione di modelli) proteico comparativo, spesso chiamato anche modelling per omologia.
Consiste nell'estrapolazione della struttura per una nuova sequenza (detta target ovvero obiettivo) dalla conoscenza preesistente sulla struttura tridimensionale (detta template ovvero stampo) di membri della stessa famiglia genetica.
Ne derivano modelli a bassa risoluzione che contengono sufficiente informazione sull'arrangiamento spaziale di residui chiave da aiutare i biologi molecolari nella pianificazione degli esperimenti.
L'assunzione alla base della modellistica per omologia è che le proteine che condividano un'elevata percentuale di residui identici (sopra il 30% di identità vi è una ragionevole sicurezza) nelle loro sequenze abbiano lo stesso fold globale, ovvero lo stesso tipo di ripiegamento strutturale, la medesima struttura di base tridimensionale [Chothia e Lesk 1986; Sander e Schneider 1991]. Spesso proteine la cui struttura si è rivelata simile presentano meno del 12% di identità di sequenza [Rost 1997].
Per modellare la struttura di una proteina sulla base delle informazioni strutturali di una proteina ad essa altamente omologa (lo stampo) si procede secondo il seguente schema:
Vengono quindi usate delle tabelle riportanti i rotameri più probabili per ogni catena laterale in relazione alla conformazione del backbone (lo scheletro di legami peptidici formato dalle parti invarianti degli aminoacidi costituenti la catena proteica).
Tutti i rotameri possibili per i residui mancanti sono analizzati con un test per ingombro sterico (ovvero vengono scartati quelli che creerebbero bump, sovrapposizioni tra gli atomi). Il rotamero più favorito è aggiunto al modello.
Vengono infine eseguite minimizzazioni energetiche con un campo di forze (descritti nel seguente capitolo) per rimuovere eventuali contatti sfavorevoli residui e per controllare le geometrie di legame modificandole se necessario.
Le minimizzazioni energetiche tendono a modificare la geometria complessiva della molecola, facendola divergere dalla struttura stampo. Si rende quindi necessario mantenere il numero di passi di minimizzazione al minimo o costringere la posizione di alcuni atomi (per esempio i carboni alfa) in ogni residuo in modo da evitare una deriva eccessiva durante i calcoli di meccanica molecolare.
La qualità di un modello è determinata da due criteri i quali definiscono la sua applicabilità:
Questi loop corrispondono quasi sempre alle parti più flessibili della struttura, come evidenziato dai loro alti fattori termici cristallografici (o dalle strutture multiple ottenute da vincoli NMR).
All'altro capo vi sono i residui del core - i meno varianti in qualunque famiglia proteica - che si ritrovano praticamente nella stessa orientazione sia nel modello che nella struttura sperimentale di controllo, mentre i residui superficiali presentano maggiore devianza.
Tutto ciò è prevedibile dato che i residui del core sono generalmente molto conservati e la loro conformazione costretta, dipendente, da quella dei residui vicinali. Per i residui superficiali queste influenze sono molto meno marcate e i pochi constraint (vincoli) sterici determinano la maggiore devianza.
La descrizione matematica completa di una molecola che includa effetti quantomeccanici e relativistici è un problema formidabile data la piccola scala di grandezza e le forti velocità.La meccanica e la dinamica molecolare sono basate su dati empirici che implicitamente incorporano tutti gli effetti quantomeccanici e relativistici.
Solitamente il punto di partenza è l?equazione di Schrödinger:
![]() HY(R,r)
= EY(R,r)
HY(R,r)
= EY(R,r)
dove H è l?Hamiltoniano del sistema, E è l?energia e Y è la funzione d?onda.
L?equazione di Schrödinger ammette soluzione solo per certi valori
di E e questi valori (detti autovalori) sono le sole energie che
il sistema può assumere. ![]() dipende dalla posizione degli elettroni (indicata globalmente con r)
e da quella dei nuclei (indicata con R).
dipende dalla posizione degli elettroni (indicata globalmente con r)
e da quella dei nuclei (indicata con R).
Quest?equazione, pur molto generale, è troppo complessa per usi pratici quindi varie approssimazioni sono adoperate.
Una prima approssimazione molto importante permette di semplificare notevolmente la descrizione. A causa delle diverse masse di nuclei ed elettroni (il rapporto ad esempio fra il peso di un elettrone e quello dell?atomo di idrogeno è dell?ordine di 10-5), si può assumere che il movimento dei nuclei sia seguito da un quasi immediato riarrangiamento degli elettroni (approssimazione di Born-Oppenheimer [1927]).
In base a questo è possibile considerare separatamente l?equazione di Schrödinger per gli elettroni e considerare l?insieme delle coordinate nucleari come dei parametri (quindi con un valore fissato). Quindi è possibile trovare l?energia dello stato fondamentale degli elettroni (che dipende dalla posizione fissata per i nuclei) risolvendo l?equazione di Schrödinger:
![]()
In questo modo ogni configurazione dei nuclei è stata associata
ad un'energia del sistema che include (implicitamente) i contributi dagli
elettroni: ![]() . Questa energia
. Questa energia ![]() può essere sostituita nell?operatore H per scrivere un'equazione
di Schrödinger che descriva la funzione d?onda per i soli nuclei e
che tenga conto degli elettroni implicitamente attraverso
può essere sostituita nell?operatore H per scrivere un'equazione
di Schrödinger che descriva la funzione d?onda per i soli nuclei e
che tenga conto degli elettroni implicitamente attraverso ![]() :
:
![]()
In linea di principio la prima delle due equazioni potrebbe essere risolta per l?energia potenziale E e quindi la seconda potrebbe venir poi risolta. Ma data la difficoltà intrinseca viene solitamente usata un?approssimazione (fit) empirica per l?equazione elettronica.
Lo studio del moto dei nuclei è denominato quantodinamica ma, poiché i nuclei sono particelle relativamente pesanti, gli effetti quantomeccanici possono essere trascurati a questo livello di approssimazione e quindi tale equazione può essere sostituita dall?equazione (classica) del moto di Newton:
![]()
Tanto la minimizzazione energetica quanto la simulazione della dinamica molecolare di un sistema a questo livello di approssimazione richiedono un'adeguata approssimazione empirica per l'energia potenziale V(R).
Il "fit" ovvero la miglior approssimazione empirica all'energia potenziale costituisce ciò che viene chiamato campo di forze. Il campo di forze definisce le coordinate usate, le equazioni che coinvolgono tali coordinate e i parametri che entrano in gioco nelle formule approssimanti l'energia potenziale.
I campi di forze comunemente usati per la descrizione e la simulazione di molecole calcolano le energie e le forze in funzione delle coordinate interne (quali distanze e angoli di legame) e delle distanze interatomiche. Questi due insiemi di dati sono usati rispettivamente per descrivere i due aspetti dell'energia potenziale: quelli legati alla struttura covalente (distanze e angoli di legame, barriere torsionali; complessivamente definiti "bond", "di legame") e quelli legati alle interazioni elettrostatiche e di van der Waals tra atomi separati da più di due legami (definiti comunemente "di non legame").
I campi di forze estrapolano i dati empirici del ristretto insieme di piccole molecole usato per la parametrizzazione al fine di simulare un ben più ampio spettro di strutture molecolari.
In altre parole, degli studi quantomeccanici accurati su piccoli sistemi permettono di ricavare la forma funzionale dell'energia i cui parametri vengono quasi sempre adattati in modo tale da riprodurre i dati sperimentali (informazioni strutturali, energie vibrazionali, barriere energetiche rotazionali, momenti dipolari) ottenuti su sistemi modello rappresentativi della classe di composti che si intende studiare.
Con una rappresentazione facilmente visualizzabile e largamente adoperata, gli atomi vengono paragonati a sfere vibranti connesse da molle.
Tale modello classico ha comunque forti limiti. Analizziamo la differenza tra un legame "classico" e uno quantomeccanico, nell?approssimazione di oscillatori armonici Figura B-11.
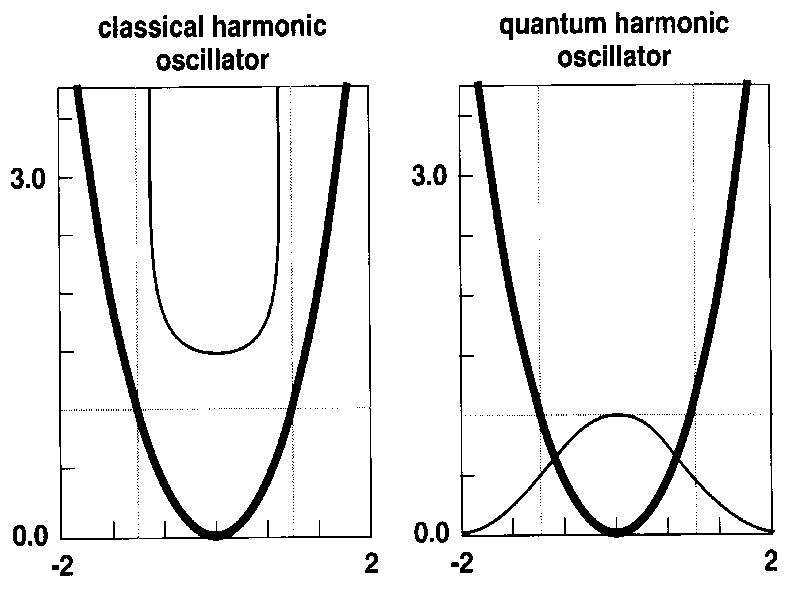 Figura
-11: Energia e probabilità di una particella classica e quantomeccanica
in un oscillatore armonico
Figura
-11: Energia e probabilità di una particella classica e quantomeccanica
in un oscillatore armonico
L'energia è indicata dalle parabole in neretto, la probabilità dalle linee sottili. L'energia totale del sistema è indicata con la linea tratteggiata orizzontale.
La probabilità classica è massima quando la particella raggiunge la sua massima energia potenziale (a velocità e quindi energia cinetica zero) e diventa nulla oltre questi punti.
La probabilità quantomeccanica è invece massima dove l'energia potenziale è minore ed esiste una certa probabilità che la particella si trovi oltre i limiti classici (indicati dalle linee tratteggiate verticali).
Sembrerebbe quindi poco ragionevole usare un approccio classico per entità ovviamente quantomeccaniche come i legami atomici. In pratica molte proprietà sperimentali, come ad esempio le frequenze vibrazionali o le strutture cristalline, possono essere riprodotte con un campo di forze classico. Questo avviene non perché i sistemi descritti si comportino classicamente ma perché il campo di forze è adattato grazie all?osservazione sperimentale e quindi include empiricamente molti degli effetti quantici.
Esempi di applicazioni che i campi forza sono impossibilitati a simulare dato l?approccio classico:
Per un esempio della forma delle equazioni che presenti in un campo di forze, si veda la trattazione sul campo CVFF (§ C.I.2.1).
L?elaborazione alla base di una simulazione biomolecolare è
il calcolo dell?energia potenziale per una certa configurazione di atomi.
Il calcolo di tale energia - e delle sue derivate rispetto alle coordinate
atomiche - fornisce l?informazione necessaria per compiere minimizzazioni,
analisi vibrazionale e simulazioni dinamiche.
Data un?equazione per l?energia potenziale e un punto di partenza, un algoritmo di minimizzazione deve determinare sia la direzione verso il minimo che la distanza da esso in quella direzione. Una buona direzione iniziale è la pendenza della derivata della funzione in quel punto. Se le derivate sono proporzionali alle coordinate, esse saranno più grandi tanto più lontani si è dal minimo.
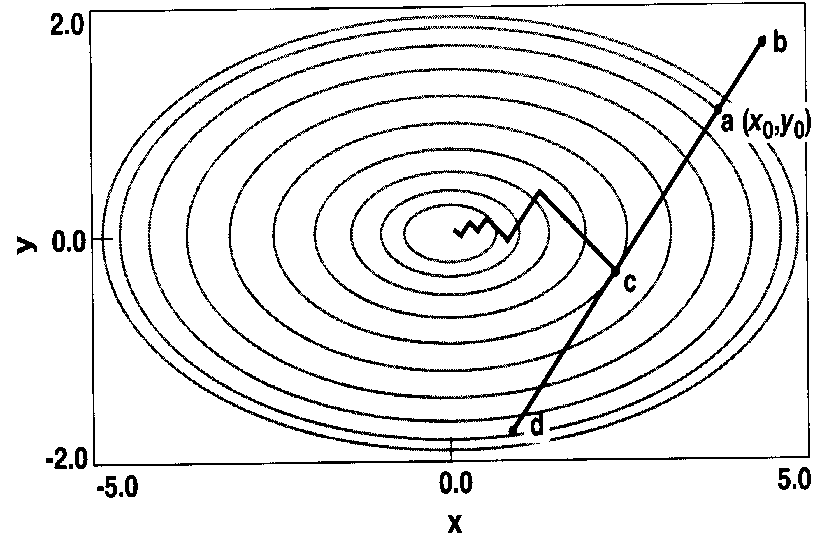
La parte più generica di un algoritmo di minimizzazione è la cosiddetta "line search" che identifica una direzione di avvicinamento al minimo e cerca il minimo energetico in questa direzione.
Gli atomi vengono quindi mossi (in genere si tratta di spostamenti molto ridotti, dell?ordine di decimi di Ångstrom) nella posizione di minimo. A questo punto si calcola nuovamente la derivata e si ripete l?operazione. Questo si farà tante volte fino a quando l?energia fra un passo e il successivo non vari più apprezzabilmente.
Una "line search" consiste quindi in una minimizzazione monodimensionale lungo un vettore direzione determinato per ogni iterazione dell?algoritmo.
Con riferimento alla Figura B-12 che mostra una superficie energetica bidimensionale, la superficie monodimensionale (la posizione del sistema nella direzione di avvicinamento al minimo) può essere espressa parametricamente in termini di a, una variabile che viene aggiustata in modo tale da minimizzare il valore della funzione E(x?,y?).
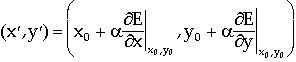
 (x?,y?) sono coordinate
lungo la linea che va dal punto (
(x?,y?) sono coordinate
lungo la linea che va dal punto (![]() )
nella direzione del gradiente in (
)
nella direzione del gradiente in (![]() ).
Il minimo lungo questa direzione, c, coincide con il punto in cui la linea
è tangente al profilo energetico (isoipse energetiche della figura).
Poiché la direzione della derivata massima è perpendicolare
alla linea di ricerca in questo punto, ogni nuova linea è ortogonale
alla precedente.
).
Il minimo lungo questa direzione, c, coincide con il punto in cui la linea
è tangente al profilo energetico (isoipse energetiche della figura).
Poiché la direzione della derivata massima è perpendicolare
alla linea di ricerca in questo punto, ogni nuova linea è ortogonale
alla precedente.
Figura -12: Linee di ricerca del minimo per una superficie bidimensionale di energia
Da notare che il vettore derivata (b-d) non punta direttamente verso il minimo. In questo vettore definito dal punto di partenza a viene ricercato il minimo (c) e da lì una nuova linea di ricerca viene tracciata.
Semplicemente minimizzando l?energia si possono individuare conformazioni stabili. Combinando le strategie di minimizzazione con l?uso di restrizioni energetiche si possono creare varie ipotesi di modellistica. Per esempio calcolare quanta energia è richiesta perché una molecola adotti una certa conformazione.
La dinamica molecolare si distingue dalla meccanica molecolare perché tiene in considerazione l?evoluzione del sistema nel tempo.
Un importante uso della dinamica molecolare è quello di esplorare lo spazio conformazionale per individuare conformazioni che siano stabili (spesso definito come conformational sampling).
L'importanza della dinamica molecolare risiede nell'essere più efficiente del metodo Montecarlo (ricerca conformazionale basata su variazioni casuali della struttura, computazionalmente proibitiva per grandi sistemi molecolari) e nel permettere il superamento delle barriere energetiche che nella semplice minimizzazione energetica non possono essere attraversate, impedendo quindi di raggiungere il vero minimo globale della struttura (cfr. § D.IV).
Una simulazione di dinamica molecolare inizia con una conformazione iniziale di atomi ed una distribuzione di velocità corrispondente ad una data temperatura, che nel tempo evolvono secondo le equazioni del moto.
Nella sua forma più semplice, una dinamica molecolare risolve l?equazione del moto di Newton:
![]()
La forza su un atomo i può essere calcolata direttamente
dal gradiente dell?energia potenziale V rispetto alle coordinate ![]() :
:
![]()
Facendo ciò si può focalizzare il calcolo su una regione o su una conformazione di interesse o si possono eseguire esperimenti computazionali.
Per orientare una simulazione vengono applicati dei vincoli che si distinguoino principalmente in due categorie: constraint e restraint. I primi sono condizioni assolute che devono essere rispettate, quali ad esempio atomi fissati nello spazio ai quali non viene permesso di muoversi. I secondi sono invece termini aggiuntivi nell?espressione dell?energia per forzare il sistema a comportarsi in un certo modo; per esempio aggiungendo un potenziale torsionale ad un certo legame si può forzare quell?angolo a tendere verso quel valore desiderato.
La differenza tra le due categorie risiede quindi nel fatto che un constraint è una restrizione assoluta imposta ai calcoli, mentre un restraint è un termine energetico che tende ad influenzare i calcoli per seguire la restrizione.
I tipi di vincoli possibili sono i seguenti:
[Clarke, 1995]
Un aminoacido che contribuisca alla stabilità o alla funzione di una proteina in un contesto di sequenza può essere superfluo o addirittura sfavorevole in un altro.
In termini evolutivi la "fitness" (convenienza) di un aminoacido in una certa posizione all'interno di una proteina dipende dagli aminoacidi presenti in altre posizioni.
Un esempio di questo potrebbe essere un residuo Aspartico in una posizione che nella struttura 3D è sepolta all'interno della proteina. Se la struttura di questa permettesse la formazione di un ponte salino tra l'acido Aspartico e una Lisina, allora ci si aspetterebbe una sua alta fitness rispetto ad altri aminoacidi. Se però la Lisina fosse sostituita da un aminoacido idrofobico, la fitness dell'Aspartico diminuirebbe drasticamente. Anche la fitness di altri aminoacidi in altre posizioni cambierebbe, ma probabilmente in modo molto meno rilevante.
Il fatto che un cambiamento aminoacidico influenzi la fitness di altri aminoacidi in un modo che riflette i "collegamenti" strutturali e funzionali suggerisce che le sequenze evolutivamente imparentate contengano le vestigia di questi effetti nella forma di coppie aminoacidiche correlate (covarianti).
Si distingue tra coppia di residui e coppia aminoacidica. Il primo termine si riferisce ad una coppia di posizioni aminoacidiche (numeri di sequenza) mentre il secondo viene usato per indicare un particolare paio di aminoacidi trovati ad una particolare coppia di residui.
Ad esempio se D è l'amminoacido alla posizione i e K l'amminoacido in posizione j, la coppia di residui è i-j, la coppia aminoacidica in i-j è D-K.
Differenti coppie aminoacidiche possono dare un contributo diverso alla covarianza totale di una coppia di residui. Continuando nell'esempio scelto, ci si aspetta che aminoacidi di carica opposta contribuiscano fortemente alla covarianza poiché i due aminoacidi hanno un significativo effetto reciproco sulla loro fitness.
In sequenze in cui il ponte salino sia sostituito da una coppia di aminoacidi idrofobici, queste coppie aminoacidiche dovrebbero contribuire meno alla covarianza della coppia di residui poiché la sostituzione di un aminoacidi idrofobico con un altro alla i-esima posizione avrebbe generalmente un effetto meno drammatico, rispetto al caso del ponte salino, sulla fitness di un secondo aminoacido idrofobico in posizione j.
Non é compito facile misurare la covarianza senza incorrere in influenze (i cosiddetti "bias") derivanti soprattutto da spinte evolutive (ad esempio alcune proteine di una famiglia con un preciso ruolo funzionale potrebbero essere state sottoposte a pressione evolutiva per ottenere un altro ruolo funzionale o strutturale) e dal campionamento (evolutivo o sperimentale, che può rivelarsi incompleto o poco rappresentativo).
Un problema nell'ottenere un insieme rappresentativo di sequenze deriva dal fatto che il campionamento delle stesse non é filogeneticamente uniforme.
Vi sono solitamente molte più sequenze derivanti dagli organismi più studiati (ad esempio Drosophila o Arabidopsis). Un'altra causa di bias nel campionamento é che spesso molte sequenze vengono trovate grazie alla similarità di sequenza con sequenze conosciute, portando ad un raggruppamento più pronunciato di sequenze molto simili di quanto sia l'effettivo insieme biologico esistente.
La via migliore per affrontare le influenze negli insiemi di sequenze non è chiara. Vari metodi sono stati proposti, basati sulla costruzione di un albero filogenetico guida [Shindyalov et al. 1994], sulla costruzione di un sottoinsieme di sequenze in modo che ognuna differisca da tutte le altre per un certo grado di identità o analogia aminoacidica [Nether 1994; Taylor et al. 1994] o abbassando il contributo di ogni sequenza in relazione alla sua similarità con le altre sequenze dell'insieme [Gobel et al. 1994]. Non ci sono forti basi teoriche per preferire una procedura all'altra.
Eliminare o pesare diversamente le sequenze necessariamente influenza il contenuto di informazione del campione e non è possibile conoscere quanto l'insieme di sequenze note differisca dall'insieme reale (o da quello teorico).
La covarianza aminoacidica (detta anche informazione mutuale; il grado con cui una coppia aminoacidica alla coppia di residui i e j mostri covarianza, co-variazione) è definita come:
dove la sommatoria è eseguita per tutte le sequenze. ai
e bj sono gli aminoacidi trovati al residuo i
e j rispettivamente, nella sequenza n; ![]() è
la probabilità che l'aminoacido ai si possa trovare
all'i-esimo residuo (considerata equivalente alla frequenza osservata
con cui il particolare aminoacido si trova al residuo i tra tutte
le sequenze) e
è
la probabilità che l'aminoacido ai si possa trovare
all'i-esimo residuo (considerata equivalente alla frequenza osservata
con cui il particolare aminoacido si trova al residuo i tra tutte
le sequenze) e![]() la probabilità
(frequenza osservata) di trovare l'aminoacido bj in posizione
j. Infine
la probabilità
(frequenza osservata) di trovare l'aminoacido bj in posizione
j. Infine ![]() rappresenta
la probabilità (frequenza osservata) di ritrovare, nella stessa
sequenza, sia l'aminoacido ai alla posizione i
che l'aminoacido bj alla posizione j.
rappresenta
la probabilità (frequenza osservata) di ritrovare, nella stessa
sequenza, sia l'aminoacido ai alla posizione i
che l'aminoacido bj alla posizione j.
Quindi ![]() ·
·![]() (il
prodotto delle probabilità individuali per ai
e bj) è la frequenza prevista per la coppia aminoacidica
ai-bj alla coppia di residui i-j
se sono indipendenti l'uno dall'altro, mentre
(il
prodotto delle probabilità individuali per ai
e bj) è la frequenza prevista per la coppia aminoacidica
ai-bj alla coppia di residui i-j
se sono indipendenti l'uno dall'altro, mentre ![]() è
la frequenza osservata per la coppia aminoacidica.
è
la frequenza osservata per la coppia aminoacidica.
Si introducono qui alcuni elementi di struttura per la loro importanza
in relazione alla presente trattazione.
[Musafia et al. 1995]
I ponti salini sono interazioni ioniche tra aminoacidi carichi di segno opposto. Occupano un ruolo molto importante nella struttura e nella funzione delle proteine.
Gli aminoacidi coinvolti sono cinque: acido Aspartico, acido Glutammico, Lisina, Arginina ed Istidina. I primi due sono acidi, con la possibilità di portare una carica negativa. I rimanenti sono basici (ammine), potenzialmente carichi positivamente. L'aminoacido Istidina è spesso non incluso nella trattazione di ponti salini perché raramente protonato e quindi carico.
Si possono definire due classi di ponti salini: semplici e complessi.
Un ponte salino semplice è un interazione ionica di non legame (o con un legame ad idrogeno) tra una singola coppia di residui aminoacidici carichi. Un ponte salino complesso unisce più di due residui (ad esempio la triade Asp-Arg-Asp) in catene proteiche singole o adiacenti.
Un'analisi di distribuzione della separazione in sequenza tra residui coinvolti in ponte salino indica che la maggior parte si colloca oltre i 7 residui di distanza, con picchi oltre i 15 (23% dei residui) e oltre i 50 aminoacidi. Un altro picco nella distribuzione si osserva a separazioni di tre e quattro aminoacidi (i+3, i+4), con un totale di quasi il 20% dei residui totali. Il 50% di questi sono all'interno di una singola a -elica. Specifiche interazioni a ponte salino stabilizzano le strutture ad a -elica come visto in esperimenti con peptidi modello [Pingchiang et al. 1992].
A livello di struttura quaternaria (ovvero di raggruppamento di più polipeptidi) si evidenzia l'importanza dei ponti salini complessi nel connettere differenti catene proteiche (nel 70% dei casi studiati, rispetto al 30% per i ponti salini semplici; situazione inversa nelle interazioni all'interno di una singola catena proteica in cui i ponti salini complessi rappresentano il 28% del totale).
La lunghezza media dell'interazione ionica (la distanza interatomica tra N delle ammine e O degli acidi coinvolti nell'interazione) è praticamente la stessa (statisticamente) per ponti salini semplici e complessi:
|
Distanza media (in Å) tra N e O carichi in ponte
salino
|
||
| Residui connessi |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
(sono esclusi da questa analisi i ponti salini coinvolgenti Istidina)
Per quanto riguarda le proteine di membrana, nonostante i fattori termodinamici sfavorevoli dovuti all'inserzione di aminoacidi carichi nell'ambiente altamente idrofobico dei fosfolipidi di membrana, le strutture disponibili mettono in luce la presenza di questi aminoacidi carichi in molte regioni transmembrana di tali proteine. Circa dieci kcal/mol sono richieste per inserire un residuo carico in una regione altamente idrofobica ma un ponte salino richiederebbe circa una kcal/mol e con alcuni ponti idrogeno aggiuntivi il ponte salino sarebbe estremamente stabile [Lee et al. 1992].
Un ponte idrogeno è un'interazione non covalente in cui un
atomo di idrogeno viene condiviso tra due altri atomi (entrambi elettronegativi,
come O ed N).
La distanza tra l'idrogeno di un'ammide e l'ossigeno di un carbonile, ad esempio, è di soli 1.9 Å e non di 2.7 Å come calcolabile dalla somma dei raggi di van der Waals. Poiché l'intero guscio elettronico dell'idrogeno (che ha un solo elettrone) è piuttosto spostato verso l'atomo a cui l'idrogeno è covalentemente legato, la repulsione dei gusci elettronici tra gli atomi coinvolti nel legame ad idrogeno è piccola e rende possibile l'avvicinamento delle cariche parziali: la grande carica parziale positiva sull'atomo di H e quella negativa sull'atomo coinvolto nel ponte idrogeno. Quest'ultimo atomo è chiamato "accettore del legame idrogeno" mentre l'atomo a cui l'idrogeno è covalentemente legato viene definito "donatore del legame idrogeno".
La più corta distanza - e più alta energia - nei ponti idrogeno, è quella tra due atomi di ossigeno (che fungano da donatore ed accettore), in particolare tra fenoli (ad esempio nell'interazione tra due Tirosine) e nell'acqua. Distanze leggermente più lunghe (ed energie minori) si ritrovano nei legami ad idrogeno tra azoto ed ossigeno ed ancor meno tra azoto ed azoto o tra azoto e zolfo.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
Una stima delle energie del legame ad idrogeno può essere ottenuta dalla misura del calore di sublimazione del ghiaccio (13 kcal/mol). Gran parte di questo calore corrisponde ai legami ad idrogeno visto che il calore di sublimazione di H2S (che forma legami ad idrogeno più deboli) è di circa 6 kcal/mol. Ci sono due ponti idrogeno per molecola di H2O e l'energia di ognuno è poco più di metà della differenza tra i calori di sublimazione, circa 4 kcal/mol. Le energie dei ponti idrogeno tra gruppi ammidici e carbonilici, frequenti negli atomi dello scheletro polipeptidico delle proteine, sono di circa 3 kcal/mol.
I legami ad idrogeno sono più forti se i tre atomi interessati sono allineati:
-Od--Hd+········Od- -Nd--Hd+········Od-
Una carica positiva ha infatti minore energia potenziale tra due cariche negative quando le tre cariche sono allineate.
I legami idrogeno sono determinanti per la struttura secondaria delle proteine e spesso molto importanti per il mantenimento della struttura terziaria.
La lunghezza media è di dieci residui aminoacidici (tre giri di elica) con minimi di 4-5 e massimi che superano i 40 residui.
Tutti i legami idrogeno in un'a -elica puntano nella stessa direzione quindi le unità peptidiche sono allineate secondo lo stesso orientamento lungo l'asse dell'elica. Poiché ogni unità peptidica possiede un momento di dipolo (derivante dalla differente polarità dei gruppi NH e C=O), questi momenti dipolari sono anch'essi allineati lungo tale asse.
L'effetto risultante è la formazione di un significativo dipolo dell'elica con una parziale carica positiva all'N-terminale (estremità amminica dell'elica) ed una parziale carica negativa all'estremità carbossilica (C-terminale).
Il lavoro di ricerca è stato svolto utilizzando stazioni
grafiche Silicon Graphics dotate di sistema operativo IRIX-SGI (Indigo:
processore "Mips R4600" con velocità di clock di 134 MHz e 64 Mb
di memoria RAM; O2: processore "Mips R500" a 180 MHz, 128 Mb RAM) e un
calcolatore Pentium II (400 MHz, 128 Mb RAM) con i sistemi operativi Linux
e Windows98.
Sono stati necessari software di grafica e simulazione molecolare per la visualizzazione, manipolazione e simulazione delle strutture proteiche e software di allineamento e studio di sequenze per lo studio delle omologie all'interno della famiglia multigenica, nonché alcuni servizi disponibili in internet (banche dati e algoritmi).
Insight [Biosym technologies 1995] è un esteso programma
di grafica e simulazione molecolare (cfr. § B.VI) studiato per vari
sistemi operativi tra cui IRIX per i computer Silicon Graphics. È
realizzato dalla Biosym Technologies/MSI.
Ha una struttura modulare che include moduli come "Biopolymer" (costruzione di molecole), "Analysis" (per l'analisi di una simulazione molecolare nel tempo) e "Discover" tramite cui avviene l'interfacciamento con il programma Discover di dinamica e meccanica molecolare (cfr. § C.I.2).
La costruzione, visualizzazione e studio di molecole sono realizzati attraverso l'uso dell'interfaccia grafica di questo programma. I comandi del programma sono divisi in vari menu, accessibili con il puntatore gestito dal mouse.
Ogni comando ha un'equivalente rappresentazione scritta, permettendo la programmazione "batch" ovvero permettendo di gestire il comportamento del programma tramite la compilazione testuale di una lista di comandi. Il programma può leggere la lista ed eseguire sequenzialmente i comandi in essa contenuti, comportandosi allo stesso modo che se fosse l'utente ad impartirli tramite l'interfaccia grafica. Questo è fondamentale per sveltire ed automatizzare alcune operazioni.
Discover [Biosym technologies 1982] è un programma di dinamica
e meccanica molecolare. Permette di calcolare energie conformazionali e
di simulare la dinamica molecolare (cfr. § B.VI.2-B.VI.4)..
Può essere quindi usato per analizzare l'energia delle possibili conformazioni di una molecola, ottimizzare una struttura dal punto di vista energetico, valutare l?effetto di perturbazioni fisiche o chimiche su un sistema, calcolare le energie libere di legame, considerando effetti entropici e di solvatazione.
Ha un suo linguaggio di programmazione batch denominato DSL (Discover Simulation Language). È prodotto dalla Biosym Technologies/MSI.
Discover dispone di quattro famiglie di campi di forze: CVFF, CFF91, ESFF e AMBER.
CVFF (Consistent Valence Forcefield) è stato parametrizzato per riprodurre proprietà di peptidi e proteine. È stato ampiamente utilizzato e può quindi essere considerato adeguato per lo studio di tali biomolecole. [Dauber-Osguthorpe et al. 1988]
AMBER è stato parametrizzato e definito solo per proteine e DNA. È stato comunque usato anche per molte altre classi di molecole, grazie all?estensione che ha ricevuto da molti autori che vi hanno aggiunto parametri per adattarlo alle loro esigenze [Weiner et al. 1984].
CFF91 è un nuovo campo di forza. È stato parametrizzato grazie ad un ampio spettro di osservazioni sperimentali ed appare più accurato di campi di forza come CVFF e AMBER. Essendo più recente non è stato così accuratamente testato come questi due e quindi non è così ben caratterizzato [Maple et al. 1994].
ESFF (Extensible Systematic Forcefield) è un nuovo tipo di campo di forze ancora in corso di sviluppo. Al contrario di altri campi di forze che privilegiano l'alta accuratezza per un limitato numero di gruppi funzionali, esso cerca di provvedere alla copertura totale della tavola periodica degli elementi. È stato pensato soprattutto per rendere possibile la simulazione di composti organometallici ma non è molto accurato nel riprodurre frequenze vibrazionali o altre proprietà come energie conformazionali. Non si basa su parametri derivati da dati sperimentali ottenuti su piccole molecole ma su parametri atomici.
Il programma Discover deve conoscere i tipi atomici di ogni atomo nel sistema da simulare per poter determinare quali parametri del campo di forze debbano essere usati. Il tipo atomico in un particolare campo di forze dipende dal gruppo chimico a cui l'atomo in questione appartiene. Ad esempio il carbonio presente nella molecola di etano ha caratteristiche diverse da quelle del carbonio presente nell'etene (a causa della diversa ibridizzazione) e pertanto i rispettivi tipi atomici sono diversi. Qualora i tipi atomici non fossero esplicitamente assegnati, il programma tenterebbe di assegnarli in base alla struttura covalente. È necessario inoltre che la struttura da simulare sia chimicamente plausibile, quindi non è possibile lasciare stati di valenza aperti, senza leganti.
Questo è il campo di forze utilizzato in questo lavoro di tesi, modificato opportunamente per adattarlo alle molecole da simulare, ovvero proteine che legano pigmenti di clorofilla. Delle modifiche apportate, necessarie per simulare la presenza dell?elemento Magnesio nelle strutture light-harvesting, si parlerà durante la discussione dei risultati (cfr. § D.III).La forma analitica dell?espressione usata in CVFF per l?energia è la seguente:
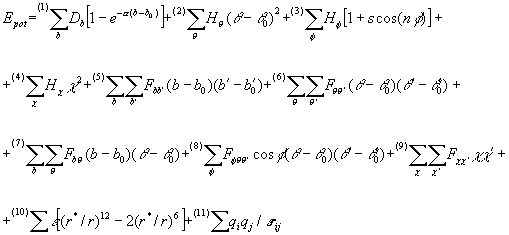
I termini contrassegnati da (1) a (4) sono comunemente definiti termini diagonali e rappresentano rispettivamente l?energia di deformazione delle distanze di legame, degli angoli di legame, degli angoli di torsione e delle interazioni non planari (angoli diedri impropri).
I termini da (5) a (9) sono anche chiamati "fuori diagonale" e rappresentano accoppiamenti tra deformazioni di coordinate interne (e.g. accoppiamento di deformazioni tra distanze di legami adiacenti).
Sono termini necessari per riprodurre correttamente le frequenze vibrazionali e, quindi, le proprietà dinamiche delle molecole.
I termini (10) e (11) descrivono le interazioni di non legame. (10) rappresenta le interazioni di van der Waals tramite una funzione di Lennard-Jones e (11) le interazioni elettrostatiche.
Si noti che qi e qj sono le cariche parziali assegnate ai singoli atomi al fine di simulare gli effetti di polarizzazione. Queste dipendono dalla struttura covalente, dall'elettronegatività che a sua volta dipende dallo stato di valenza.
La costante dielettrica e è stata posta - nelle simulazioni eseguite - uguale a 4.00, valore leggermente superiore al valore tipico per gli alcani (che va da 2 a 4), per simulare l'interno apolare della membrana.
Il campo potrebbe essere provvisto di funzioni particolari per simulare i legami idrogeno, ma questi sono una naturale conseguenza dei parametri elettrostatici e di van der Waals e l?introduzione di altre funzioni non migliorerebbe la bontà di CVFF in relazione ai dati sperimentali [Hagler et al. 1979].
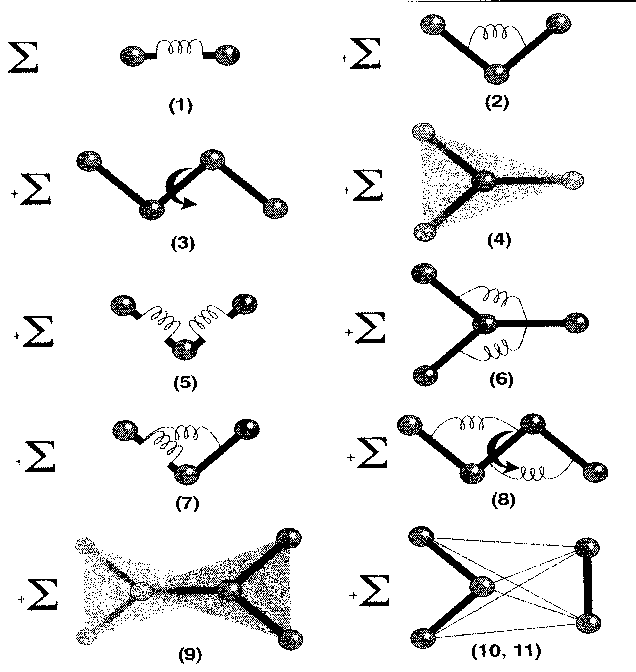
Figura -1: Visualizzazione grafica dei termini dell?equazione adottata da CVFF
WhatIf [Vriend G. 1990] è un altro programma di modellistica
molecolare (cfr. § B.VI) utilizzato.
Il programma è in grado di visualizzare contatti impropri tra gli atomi (ingombri sterici), correggerli modificando la conformazione degli atomi coinvolti (ad esempio mediante rotazioni delle catene laterali degli aminoacidi) e mostrare graficamente i cambiamenti avvenuti nella struttura.
Il programma inoltre è capace di creare mappe di densità elettronica, di colorare i residui in base alle loro caratteristiche (carica, idrofobicità, tipo di residuo) ed ogni operazione può tener conto delle simmetrie cristallografiche.
È disponibile per i sistemi operativi di tipo UNIX ma esiste anche una versione per MS-DOS. L'indirizzo internet della pagina dedicata a questo programma è http://swift.embl-heidelberg.de/whatif.
In particolare questo programma è servito in questo lavoro di tesi per la ricostruzione della struttura (posizionamento delle catene laterali), per il modelling per omologia delle antenne minori e per l'applicazione delle simmetrie cristallografiche al monomero di LHC II per ottenere la struttura del trimero.
Software per ambiente Windows, fornisce all?utente la possibilità
di visualizzare e manipolare strutture molecolari. Le immagini di proteine
che compaiono in questa tesi sono state realizzate principalmente con questo
software in combinazione con un programma di ray-tracing POVray
(http://www.povray.org). Swiss-PDB Viewer è disponibile presso:
http://www.expasy.ch/spdbv/mainpage.html
Per la stima delle cariche parziali atomiche (cfr. § C.I.2.1)
si è utilizzato un algoritmo dovuto a Gasteiger e Marsili [1980]
basato sul concetto di elettronegatività. Proposta dapprima da Pauling
e Yost [1932] per spiegare le differenze di energie di legame omo- ed etero-nucleare,
il concetto di elettronegatività è stato poi esteso dal livello
atomico al livello di stato di valenza. In particolare la seguente definizione
quantitativa di elettronegatività in termini di energia di ionizzazione
I ed affinità elettronica E venne proposta da Mulliken
[1934]:
![]()
dove n si riferisce allo stato di valenza dell'atomo piuttosto che all'atomo in sé.
Hinze e Jaffé [1962, 1963], seguendo questa linea, calcolarono - a partire dalle energie di ionizzazione e dalle affinità elettroniche dello stato atomico più stabile e dalle energie di promozione allo stato di valenza - le energie di ionizzazione, le affinità elettroniche e le elettronegatività di un grande numero di stati di valenza per numerosi atomi.
Chiaramente l'elettronegatività varia con lo stato di occupazione degli orbitali atomici.
Per tenere conto di questo fatto e per risolvere i problemi legati ad una semplice equalizzazione delle elettronegatività degli atomi in un certo stato di valenza in una molecola, Gasteiger e Marsili proposero il seguente algoritmo iterativo di parziale equalizzazione di elettronegatività orbitale (PEOE).
Al primo passo si assegna ad ogni atomo la sua carica formale. Al passo successivo (i-esimo) la carica viene ridistribuita fra due atomi legati covalentemente secondo l'equazione:
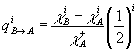
dove ![]() è la carica
parziale trasferita al passo i-esimo dall'atomo B all'atomo A e
i valori di cB e cA
dipendono dalla carica parziale sull'atomo secondo l'equazione:
è la carica
parziale trasferita al passo i-esimo dall'atomo B all'atomo A e
i valori di cB e cA
dipendono dalla carica parziale sull'atomo secondo l'equazione:
cB=a+bQB+cQB2
dove QB è la carica parziale sull'atomo B e dove i coefficienti a, b e c sono legati ai valori tabulati di Hinze e Jaffé. In particolare:
![]()
![]()
![]()
Generalmente l'algoritmo converge entro cinque cicli.
MaxSprout [Holm et al. 1991] è un algoritmo accessibile in
internet per la generazione delle coordinate di catene laterali aminoacidiche
a partire dalla traccia polipeptidica ovvero dalle coordinate dei carboni
alfa.
La conformazione delle catene laterali è ottimizzata nella scelta dei possibili rotameri usando una funzione approssimata di energia potenziale per evitare sovrapposizioni, ingombri sterici tra gli atomi costituenti le catene.
Per la ricostruzione del backbone (lo scheletro polipeptidico) viene utilizzato un database ridotto di strutture proteiche, in modo da ricercare dei frammenti proteici che si adattino meglio alle coordinate della traccia Ca . I frammenti migliori vengono sovrapposti alla traccia cercando di minimizzare eventuali discontinuità nei punti di unione tra frammenti diversi.
MaxSprout legge la traccia in formato PDB e restituisce le coordinate nello stesso formato.
Il database usato per la ricostruzione del backbone si avvale della struttura di alcune proteine i cui codici PDB sono i seguenti:
| 9ins | 1lh1 | 1mbo | 1nxb | 2ovo | 8pti | 2ptn | 1rei |
| 1rhd | 3sga | 2sns | 4tln | 1ppt | 4cac | 2pab | 351c |
| 2act | 3app | 2aza | 3b5c | 1bp2 | 2c2c | 5cpa | 4cpv |
| 1crn | 3cyt | 3dfr | 1ecd | 2fb4 | 1fdx | 4fxn | 1hip |
La libreria di rotameri per le catene laterali contiene le configurazioni atomiche più frequenti e favorevoli per i diversi residui aminoacidici.
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
È disponibile all'indirizzo http://www2.ebi.ac.uk/dali/maxsprout
Fornisce informazioni sulle piccole molecole di tipo non proteico che si trovano normalmente associate (covalentemente o non covalentemente) con le proteine e che siano presenti nei file di struttura presenti nella banca dati PDB (cfr. § C.II.1).
Le informazioni disponibili per ogni composto sono di vario tipo:
Vari database di sequenze o strutture (cfr. § B.V.1)
sono stati utilizzati:
SRS: Sequence Retrieval System, gateway internet per il recupero di sequenze e l?interrogazione automatica di vari database. http://srs.ebi.ac.uk:5000
SwissProt: Database di sequenze aminoacidiche. http://www.expasy.ch/sprot/sprot-top.html
TREMBL: Translated EMBL, database di sequenze aminoacidiche tradotte da sequenze genetiche presenti in EMBL. http://www.embl-heidelberg.de/AnoDb/Protseq/Trembl/
PDB: Protein Data Bank, database contenente le strutture risolte per cristallografia a raggi X o NMR. http://www.rcsb.org
http://ibc.wustl.edu/msa/clustal.cgi
Anche Macaw [Schuler et al. 1991] è un programma per l'allineamento
multiplo ma è soprattutto uno strumento grafico che aiuta l'utente
a vedere i vari possibili allineamenti e decidere quali siano da tenere,
ovvero quali abbiano il più alto significato biologico o siano più
vicini all'obiettivo perseguito dall'utente.
Incorpora un algoritmo di ricerca dei blocchi omologhi, ma si limita ad indicarli senza aprire alcun gap. È compito dell'utente quindi scegliere tra i possibili blocchi e collegarli, grazie all'interfaccia grafica.
Per Macintosh e Windows. Disponibile al sito dell?NCBI (National Center for Biotechnology Information):
ftp://ncbi.nlm.nih.gov/pub/macaw
Accetta sequenze aminoacidiche in vari formati, restituendo molti tipi di predizioni strutturali (cfr. § B.V.3) tra cui:
PHYLIP (PHYLogeny Inference Package) [Felsenstein 1989] raggruppa
sotto il suo nome una collezione di programmi per la filogenesi (alberi
evolutivi).
È disponibile in internet (http://evolution.genetics.washington.edu/phylip.html) e funziona su varie piattaforme (UNIX, Windows, Macintosh, DOS).
I tipi di dati che PHYLIP riconosce e può elaborare includono sequenze (genetiche o aminoacidiche), frequenze geniche, siti di restrizione, matrici di distanza.
Ad esempio:
|
ETAGP ETVGP ESVGP ESVGP |
P1,E: 1
P2,T: 0.5 P2,S: 0.5 |
P3,A: 0.25
P3,V: 0.75 P4,G: 1 P5,P: 1 |
Per definizione, la somma delle probabilità per ogni residuo ad una data posizione dev'essere 1.
Se si dispone di una struttura, o anche delle sole coordinate dei carboni alfa, questa può essere usata per creare una maschera strutturale usabile da AliAna.
Una maschera strutturale è una matrice bidimensionale le cui celle rappresentano tutte le possibili combinazioni a due a due di aminoacidi costituenti la proteina di cui si possiede la struttura. Ogni cella contiene la misura della distanza (in Å) intercorrente tra i Ca dei due aminoacidi.
Questa matrice viene letta dal programma AliAna e utilizzata per filtrare le informazioni.
Un cutoff (impostabile dall'utente o fissato a 13 Å) definisce quali coppie di aminoacidi si trovino (nella struttura) a meno di una certa distanza (la distanza di cutoff strutturale, appunto).
Ad esempio nella procedura di individuazione dei possibili ponti disolfuro, saranno enumerati solo quelli che soddisfino il vincolo strutturale ovvero non saranno tenuti in considerazione quelli che potrebbero formarsi tra aminoacidi che nella struttura 3D si trovano a più di 13 (e.g.) Å di distanza (più precisamente: aminoacidi i cui Ca si trovino ad una distanza maggiore di quella definita dal cutoff strutturale).
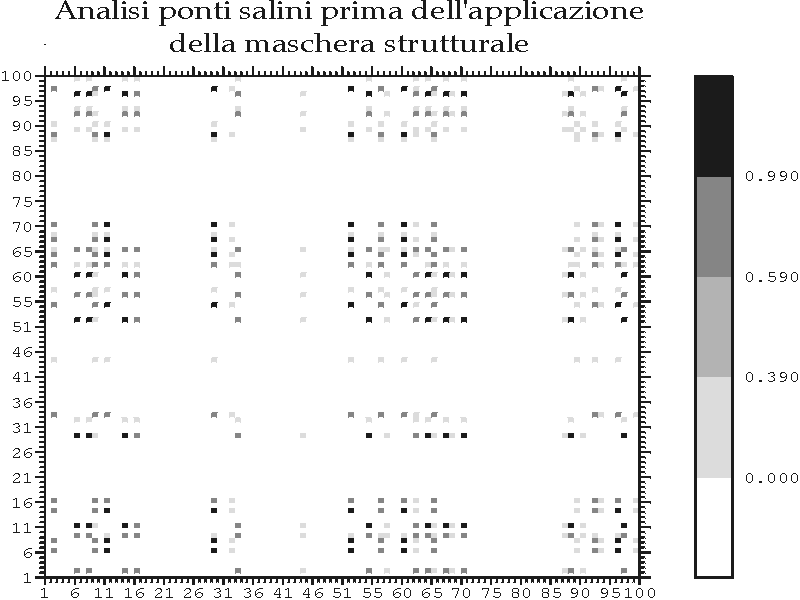
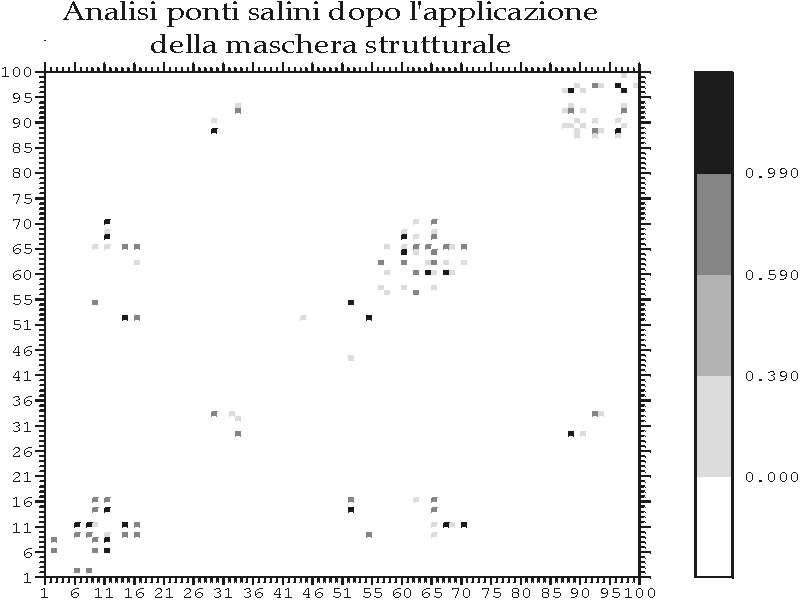 Figura
-2: Esempio di applicazione della maschera strutturale nella ricerca di
possibili ponti salini
Figura
-2: Esempio di applicazione della maschera strutturale nella ricerca di
possibili ponti salini
Nei due assi compaiono le posizioni dell'allineamento usato mentre il codice colore si riferisce alla ricorrenza relativa con cui il possibile ponte salino è osservato
La disponibilità di una maschera strutturale (anche se incompleta) è di enorme aiuto per l'analisi dei dati che il programma AliAna fornisce.
Senza di essa molti dati importanti sarebbero sommersi dai dati strutturalmente inconsistenti e sarebbe quindi più difficile "distinguere il segnale dal rumore".
Per un applicazione meno rigida del cutoff strutturale (finora descritto come dicotomico: distinzione netta tra aminoacidi con possibilità di interazione e aminoacidi senza tale possibilità) si può optare per il passaggio da una maschera strutturale binaria (in cui la misura della distanza tra una coppia di aminoacidi può essere solo minore o maggiore del cutoff strutturale) ad una maschera graduale.
In tal caso il dato per residui a distanze superiori al cutoff verrebbe moltiplicato per una funzione che varia in maniera graduale da 1 (alla distanza di cutoff) a 0 per la massima distanza ammessa per l'interazione.
Un'altra caratteristica importante di AliAna è la sua possibilità di considerare classi di aminoacidi, ovvero di raggruppare aminoacidi con stesse proprietà fisiche o chimiche e considerarli come un unico nuovo tipo aminoacidico.
Con la stessa ideologia alla base delle matrici di sostituzione (cfr. § B.V.2.1), anche qui viene sfruttata la possibilità di considerare le analogie oltre alle identità aminoacidiche. Quindi, considerando ad esempio le caratteristiche di idrofobicità, la sostituzione di una Val con una Ala non verrebbe conteggiata come contribuente alla covarianza mentre lo verrebbe la sostituzione di una Val con un Glu.
Questo può essere sfruttato per indagare più accuratamente alcuni aspetti della conservazione evolutiva all'interno della famiglia genica, per il mantenimento e di una precisa struttura proteica e di alcuni tipi aminoacidici chiave (ad esempio la presenza di aromatici in una particolare zona di sequenza o di struttura).
Il programma prevede 5 tipi di classificazioni aminoacidiche ma lascia all'utente anche la possibilità di definire le proprie classi aminoacidiche in modo da consentire un uso personalizzato.
Le cinque classificazioni predefinite sono:
Per la visualizzazione grafica dell'analisi di covarianza e dell'analisi di possibili ponti salini ci si può avvalere di vari programmi che disegnino grafici a partire da matrici quadrate (quali appunto quelle restituite da AliAna). Le immagini che compaiono in questo lavoro di tesi sono prodotte con il programma XFARBE [Preusser 1989].
Il primo passo da compiersi per lo studio della proteina in esame
consiste nella ricerca delle sequenze aminoacidiche presentanti omologia
con la proteina oggetto di studio per creare un proprio database
su cui lavorare.
Le banche dati usate sono TREMBL e SwissProt (cfr. § C.II.1). Inserendo parole chiave (keyword) essi restituiscono tutte le sequenze che le contengono (nell'header ovvero nell'intestazione e commento apposti alla sequenza, sia essa aminoacidica o genetica).
Le keyword usate per la ricerca sono quindi: ELIP (Early Light-Induced Protein), LHC (Light Harvesting Complex), CP29, CP26, CP24.
Questo tipo di ricerca produce un numero sovrabbondante di sequenze, che devono essere filtrate per ottenere ciò che effettivamente interessa.
Infatti, date le parole chiave utilizzate, sono state selezionate anche proteine di procarioti ed animali che devono essere scartate (ad esempio tale ricerca restituisce una proteina chiamata "CP24_Human" che chiaramente esula dal campo di interesse).
Vengono anche eliminate le proteine del PS I. Si è preferito infatti focalizzare l'attenzione sulla famiglia multigenica dei geni Lhcb (Lhcb1-6) ovvero su LHC II e le antenne minori CP 29, CP 26 e CP 24 anche se le proteine codificate dai geni Lhca (proteine del complesso antenna LHC I appartenente al PS I) presentano parentela con i prodotti dei geni Lhcb (come evidenziato in seguito, cfr. § D.V.3, e in Green et al. [1991]).
Le sequenze complete di proteine appartenenti alla famiglia Lhcb trovate
all'inizio del lavoro di tesi sono:
|
|
|
|
ORGANISMO (in corsivo se alga verde) |
|
|
|
|
Lycopersicon esculentum |
|
|
|
|
Lycopersicon esculentum |
|
|
|
Spinacia oleracea | |
|
|
|
|
Zea mays |
|
|
|
|
Brassica juncea |
|
|
|
|
Pinus sylvestris |
|
|
|
|
Zea mays |
|
|
|
|
Zea mays |
|
|
|
|
Lycopersicon esculentum |
|
|
|
Hordeum vulgare | |
|
|
|
|
Arabidopsis thaliana |
|
|
|
|
Zea mays |
|
|
|
|
Zea mays |
|
|
|
Amaranthus hypochondriacus | |
|
|
|
Arabidopsis thaliana | |
|
|
|
|
Arabidopsis thaliana |
|
|
|
Chlamydomonas moewusii | |
|
|
|
|
Chlamydomonas reinhardtii |
|
|
|
Cucumis sativus | |
|
|
|
Cucumis sativus | |
|
|
|
Dunaliella salina | |
|
|
|
Dunaliella salina | |
|
|
|
Dunaliella tertiolecta | |
|
|
|
Glycine max | |
|
|
|
|
Glycine max |
|
|
|
|
Glycine max |
|
|
|
|
Glycine max |
|
|
|
|
Hordeum vulgare |
|
|
|
Lemna gibba | |
|
|
|
|
Lycopersicon esculentum |
|
|
|
|
Lycopersicon esculentum |
|
|
|
|
Lycopersicon esculentum |
|
|
|
|
Lycopersicon esculentum |
|
|
|
|
Malus domestica |
|
|
|
|
Nicotiana plumbaginifolia |
|
|
|
|
Nicotiana plumbaginifolia |
|
|
|
|
Nicotiana tabacum |
|
|
|
|
Nicotiana tabacum |
|
|
|
|
Nicotiana tabacum |
|
|
|
|
Nicotiana tabacum |
|
|
|
|
Nicotiana tabacum |
|
|
|
|
Nicotiana tabacum |
|
|
|
|
Oryza sativa |
|
|
|
|
Oryza sativa |
|
|
|
Oryza sativa | |
|
|
|
|
Petunia sp. |
|
|
|
|
Petunia sp. |
|
|
|
|
Petunia sp. |
|
|
|
|
Petunia sp. |
|
|
|
|
Petunia sp. |
|
|
|
|
Petunia sp. |
|
|
|
Physcomitrella patens | |
|
|
|
Pinus thunbergii | |
|
|
|
|
Pisum sativum |
|
|
|
|
Pisum sativum |
|
|
|
|
Pisum sativum |
|
|
|
|
Polystichum munitum |
|
|
|
|
Sinapis alba |
|
|
|
Spinacia oleracea | |
|
|
|
Triticum aestivum | |
|
|
|
|
Zea mays |
|
|
|
Zea mays | |
|
|
|
|
Zea mays |
|
|
|
|
Zea mays |
|
|
|
|
Gossypium hirsutum |
|
|
|
|
Pinus sylvestris |
|
|
|
|
Pinus sylvestris |
|
|
|
|
Pisum sativum |
|
|
|
Pyrus pyrifolia | |
|
|
|
|
Hordeum vulgare |
|
|
|
|
Lycopersicon esculentum |
|
|
|
|
Pisum sativum |
Come si può vedere nella tabella, vi sono molti dati mancanti e diverse nomenclature geniche.
CB22_PEA (indicata in grassetto nella precedente tabella) è la sequenza della proteina cristallizzata da Kühlbrandt, LHC II di pisello, principale oggetto di indagine e riferimento per questo studio.
Navigando tra i database di sequenze è facile incappare in errori di attribuzione ed infatti ne è qui riportato uno: la sequenza denominata "Q40039" è classificata come "CP29 di Hordeum vulgare" mentre ad un'analisi di sequenza risulta essere con ogni probabilità una "CP26" (altissima omologia con altre CP26, scarsa con le CP29).
Inoltre molte sequenze classificate di tipo I (ovvero codificate da geni Lhcb1) sono molto probabilmente del tipo II (ovvero codificate dalla classe genica Lhcb2) come è possibile dedurre dall'albero filogenetico (cfr. § D.V.3) ovvero dal grado di omologia.
L'insieme delle sequenze così ottenuto può essere fonte di molte informazioni sull'intera famiglia multigenica anche se pecca di un campionamento sbilanciato a favore delle LHC II tipo I sia per la presenza di copie multiple di questa proteina sul genoma delle piante sia per il probabile uso di sonde mirate a Lhcb1 per la ricerca di geni codificanti per proteine dello stesso tipo.
Per gli studi di sequenza effettuati sulla famiglia, cfr. § D.V.
La ricostruzione delle catene laterali è stata ottenuta in due modi diversi:
Né Maxsprout né WhatIf riconoscono la presenza dei pigmenti quindi non ne tengono conto per l'assegnamento delle catene laterali.
Si creano in questo modo dei "bump" ovvero delle sovrapposizioni tra le catene laterali e i pigmenti.
La funzione Autorotamer del programma Insight risolve questo, usando una sua libreria di rotameri per riconfigurare le catene laterali in funzione dei bump presenti.
In realtà la disposizione così ottenuta delle catene laterali non rappresenta un modello sufficientemente plausibile. Due sono i motivi che lo rivelano:
Per poter eseguire simulazioni dinamiche e minimizzazioni energetiche (utilizzando i software Insight e Discover) è necessario avere una struttura che non presenti problemi nell'assegnazione dei tipi atomici (cfr. § C.I.2). L'assegnazione è infatti automatica solo per (parti di) molecole conosciute, presenti nelle librerie di questi programmi.
La prima difficoltà incontrata nell'assegnazione corretta di tipi atomici e cariche parziali è rappresentato dalle estremità delle eliche transmembrana. La struttura cristallografica della proteina è infatti costituita solo dalle zone degli aminoacidi
Le estremità "N-terminali" dei singoli frammenti strutturali sono state "protette" (terminate) da un gruppo acilico (-CO-CH3); le porzioni "C-terminali" sono state protette da un gruppo amidico secondario (-NH-CH3).
Questi dovrebbero mimare - almeno in maniera minima - la continuazione della catena e non dare problemi nella successiva applicazione di un campo di forze.
Gli anelli tetrapirrolici delle clorofille sono stati ricostruiti, tenendo in considerazione i legami doppi coniugati degli anelli. Non avendo alcuna informazione riguardo alla corretta orientazione si è deciso di non aggiungere il V anello mancante né la catena fitolica poiché vi sarebbero 8 diverse possibilità e una scelta incorretta minerebbe la significatività delle simulazioni molecolari che conterrebbero informazioni errate. Solo un metile è stato fornito ad ognuna delle 8 posizioni, come appropriato per la reale struttura di questi pigmenti.
 Le clorofille presenti nella
struttura ricostruita sono quindi oggetti simmetrici con un asse di simmetria
"principale", quattro assi di simmetria binaria (perpendicolari a due a
due) ed un piano di simmetria:
Le clorofille presenti nella
struttura ricostruita sono quindi oggetti simmetrici con un asse di simmetria
"principale", quattro assi di simmetria binaria (perpendicolari a due a
due) ed un piano di simmetria:
Figura -1: Struttura delle clorofille inserite nella struttura ricostruita
In realtà il Mg centrale è penta-coordinato e si trova leggermente spostato fuori dal piano macrociclico (da 0.3 a 0.5 Å) formando quindi una piramide a base quadrata; base formata dai quattro atomi di azoto del tetrapirrolo.
Figura -2: Rappresentazione schematica della struttura degli atomi Mg e N nella clorofilla
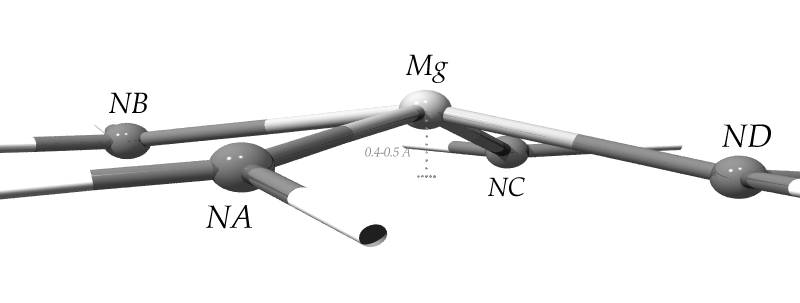
La dislocazione del magnesio, nelle strutture contenenti clorofilla, è sempre verso il lato del piano macrociclico dal quale il metallo è legato alla proteina.
L'assegnazione dei tipi atomici e delle cariche parziali è
compiuta automaticamente dal programma Insight solo per le parti polipeptidiche
della struttura.
Ma il programma non può compiere l'assegnazione automatica anche per le clorofille. Queste contengono infatti un atomo di Mg covalentemente legato con atomi di azoto nell?anello porfirinico, ben diversi dallo ione singolo Mg++ riconosciuto dal programma (ovvero riconosciuto dal campo di forze CVFF).
Per permettere la simulazione è stato quindi necessario estendere le funzionalità dei programmi a nostra disposizione, insegnando loro come trattare le clorofille: informazioni sulla distribuzione di carica parziale, sull'assegnazione dei tipi atomici e dati relativi alle caratteristiche fisico-chimiche del pigmento clorofilla.
Per l'assegnazione della carica parziale ad ogni atomo dei pigmenti
si sono sfruttati due diversi algoritmi: quello contenuto nel programma
Insight (e derivante da tipi atomici e cariche pre-assegnati per il residuo
porfirinico) e quello di un programma basato sull?algoritmo di Gasteiger
e Marsili [Gasteiger e Marsili, 1980] (cfr. § C.I.5).
 I due algoritmi impiegati
hanno portato ad una diversa distribuzione delle cariche:
I due algoritmi impiegati
hanno portato ad una diversa distribuzione delle cariche:
Insight Gast-Mars
Figura -3: Diversità nella distribuzione di carica data dagli algoritmi di Insight e Gast-Mars
A questo punto è stato operato un ragionevole compromesso tra le due distribuzioni, privilegiando quella tabulata nel residuo porfirinico di Insight per l'anello esterno ma preferendo quella originata da Gast-Mars per la parte relativa alle interazioni magnesio-azoti per alterare il meno possibile una singola componente di un campo di forze in cui l'insieme delle componenti è tarato per riprodurre dati sperimentali.
Inoltre il modello di Gasteiger e Marsili è accurato ed applicabile per sistemi a legami s e p non coniugati e quindi anche per questo motivo la distribuzione di carica operata da Insight sulla parte esterna dell?anello, ricca di doppi legami coniugati, è stata preferita.
Per simulare la delocalizzazione pressoché uniforme della carica data dalla natura risonante degli anelli, la distribuzione è stata resa uniforme e simmetrica, mediando la distribuzione di diverse strutture topologiche di risonanza.
La distribuzione di carica definitiva è indicata nella seguente figura:
Figura -4: Distribuzione di carica adottata per le clorofille della struttura ricostruita
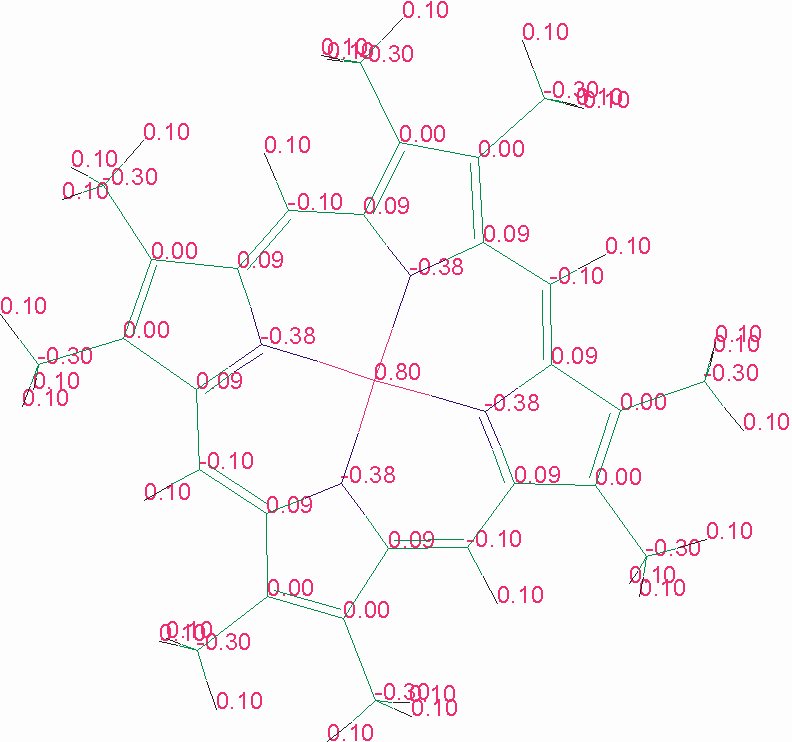
Le nuove clorofille ricostruite (leggermente diverse rispetto a quelle presenti nei dati cristallografici originali a seguito della minimizzazione) e portanti la definitiva distribuzione di carica sono state posizionate nelle corrette locazioni all'interno della struttura PDB originale.
Lo stesso è stato fatto per le due xantofille presenti (ricostruite anch'essi con gli anelli contenenti ossigeno).
Dopo uno studio attento della sintassi dei file descriventi
il campo di forze CVFF sono stati definiti tre nuovi tipi atomici: Mg',
np', np".
Il file principale per la descrizione del campo di forze ha nome cvff.frc ed è un file di testo contenente l?enunciazione di tutti i tipi atomici, e i corrispondenti parametri nel campo di forze (cfr. § F.I).
Mg' è definito come atomo di magnesio contenuto in un anello tetrapirrolico di clorofilla. Sono stati inseriti quindi tutti i parametri relativi alle interazioni necessarie per la simulazione, specialmente in relazione agli atomi di azoto.
np? e np" si riferiscono all'atomo di azoto planare sp2 aromatico. La definizione di questi due nuovi tipi è necessaria per una descrizione accurata dell?anello tetrapirrolico.
I tipi atomici np' e np" sono stati assegnati alternativamente ai quattro atomi di azoto che circondano il Mg nella clorofilla. La disposizione è quindi di tipi atomici uguali a due a due nelle estremità dei bracci della "croce" formata dagli atomi di azoto.
Schematicamente:
N
|
N---Mg---N Gli atomi al centro dell?anello della clorofilla
|
N
np'
|
np"--Mg'--np" Assegnazione dei tipi atomici
|
np'
Questo ha permesso di definire separatamente gli angoli come np'-Mg'-np' (e l?equivalente np"-Mg'-np") da quelli come np'-Mg'-np". Entrambi gli angoli hanno il magnesio come vertice ma i due estremi sono nel primo caso a estremità opposte della "croce" e nel secondo sono invece ortogonali.
Alcuni dati necessari per parametrizzare il tipo atomico Mg? sono tabulati (ad esempio la massa atomica), per gli altri si è fatto ricorso ad una stima a partire dalle strutture depositate in PDB.
Di seguito viene riportata la lista di tali strutture, dove sono indicati il codice di accesso PDB, il nome depositato per la proteina e l'organismo da cui la proteina proviene.
|
|
Nome | Organismo |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
|
|
Bacteriochlorophyll A protein | Chlorobium tepidum |
|
|
Light Harvesting Complex | Rhodopseudomonas acidophila |
|
|
Light Harvesting Complex II | Rhodospirillum molischianum |
|
|
Photosynthetic Reaction Center mutant | Rhodobacter sphaeroides |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
|
|
Peridinin Chlorophyll protein | Amphidinium carterae |
|
|
Photosynthetic Reaction Center & Core Antenna System | Synechococcus elongatus |
|
|
Photosynthetic Reaction Center | Rhodopseudomonas viridis |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
|
|
Bacteriochlorophyll A protein | Prosthecochloris aestuarii |
|
|
Photosynthetic Reaction Center | Rhodobacter sphaeroides |
Da queste strutture sono state estratte le coordinate relative alle molecole di clorofilla presenti, per un totale di 180 pigmenti.
Queste sono state analizzate (creando opportuni strumenti software) per ottenere i parametri necessari, sia in termini di distanze atomiche che in termini di angoli di legame.
Per ogni molecola di clorofilla sono stati estratti i valori delle distanze atomiche (rilevanti quelle tra Mg e gli atomi di azoto del macrociclo) e degli angoli di legame riguardanti l?anello tetrapirrolico.
I dati sono stati poi posti in grafico in forma di istogrammi di distribuzione. L?insieme delle misure per ogni caratteristica ricercata (ad esempio lunghezza di legame, angolo di legame) è stato diviso in intervalli e per ogni intervallo è stata calcolata la frequenza, vale a dire il numero di misure appartenenti ad un dato intervallo.
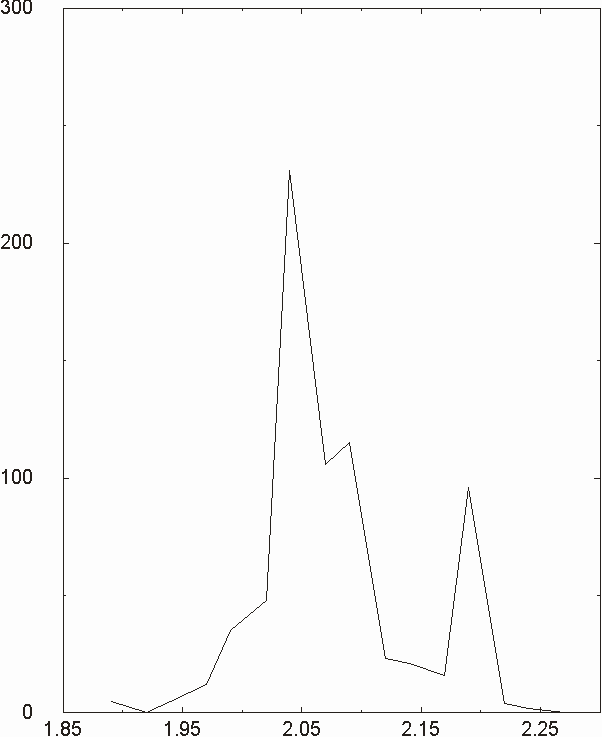 Di seguito vengono riportate
le informazioni relative ai dati (numero totale dati, valore minimo, valore
massimo e media aritmetica) e gli istogrammi derivati per ogni tipo di
angolo e di misura atomica considerati (in ascissa distanze o angoli, in
ordinata la ricorrenza dei dati).
Di seguito vengono riportate
le informazioni relative ai dati (numero totale dati, valore minimo, valore
massimo e media aritmetica) e gli istogrammi derivati per ogni tipo di
angolo e di misura atomica considerati (in ascissa distanze o angoli, in
ordinata la ricorrenza dei dati).
|
|
|
|
|
|
|
|
|
|
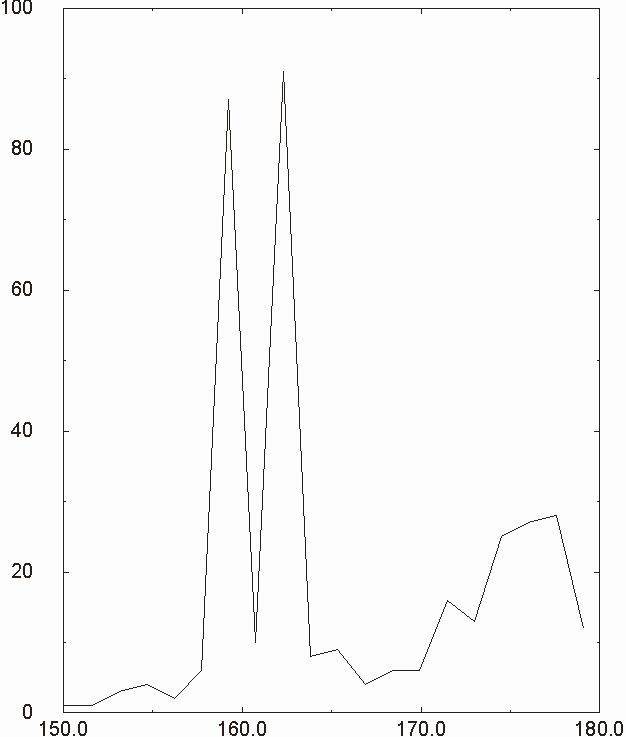 angoli N-Mg-N
angoli N-Mg-N|
|
|
|
|
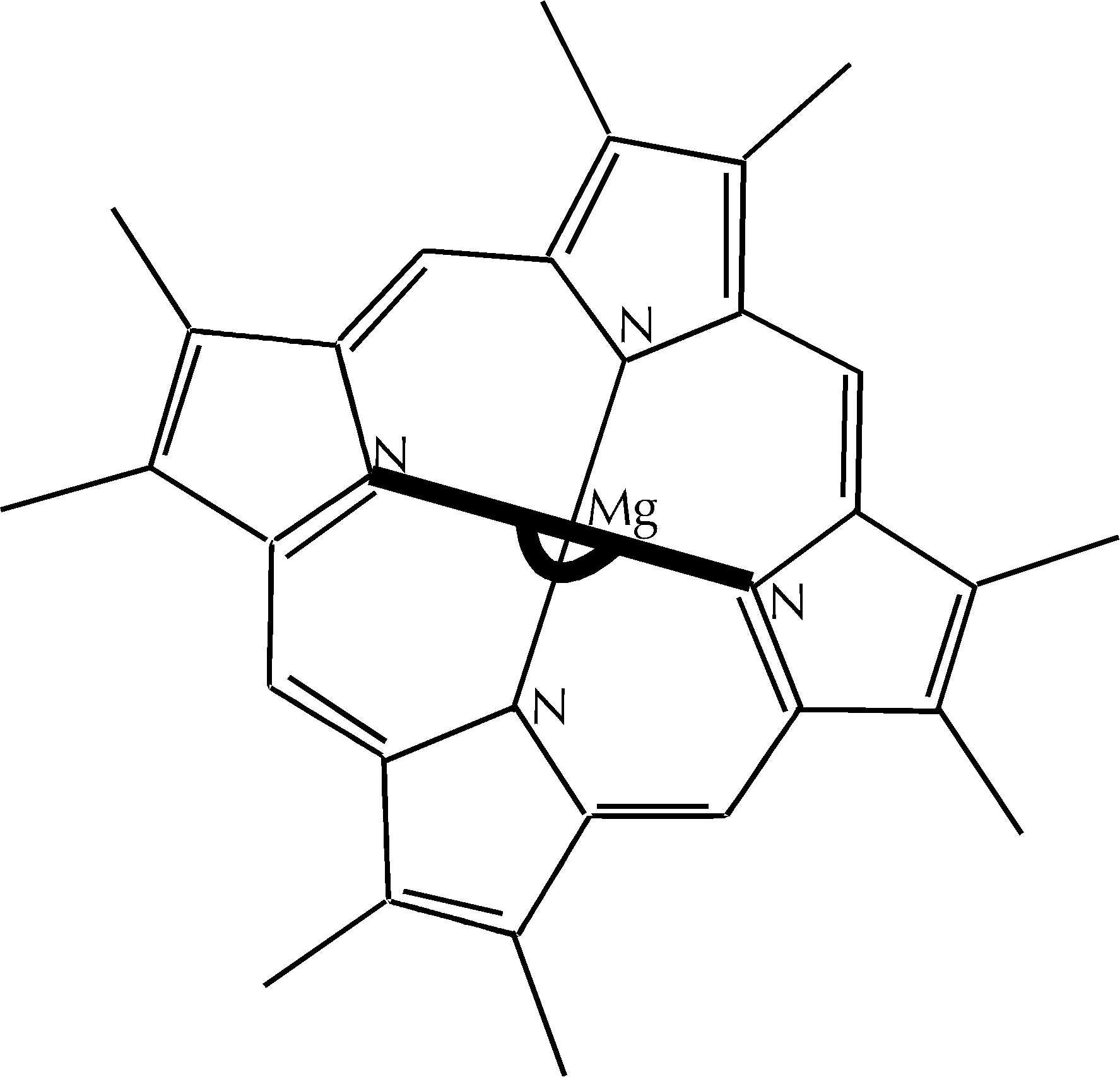 360 360 |
|
|
|
Figura -6: Rappresentazione dell'angolo N-Mg-N
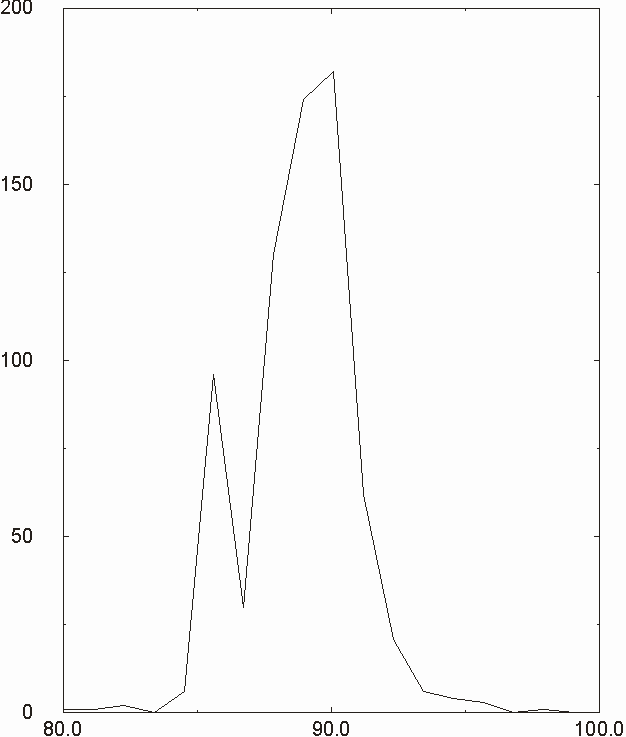 angoli N-Mg-N
angoli N-Mg-N|
|
|
|
|
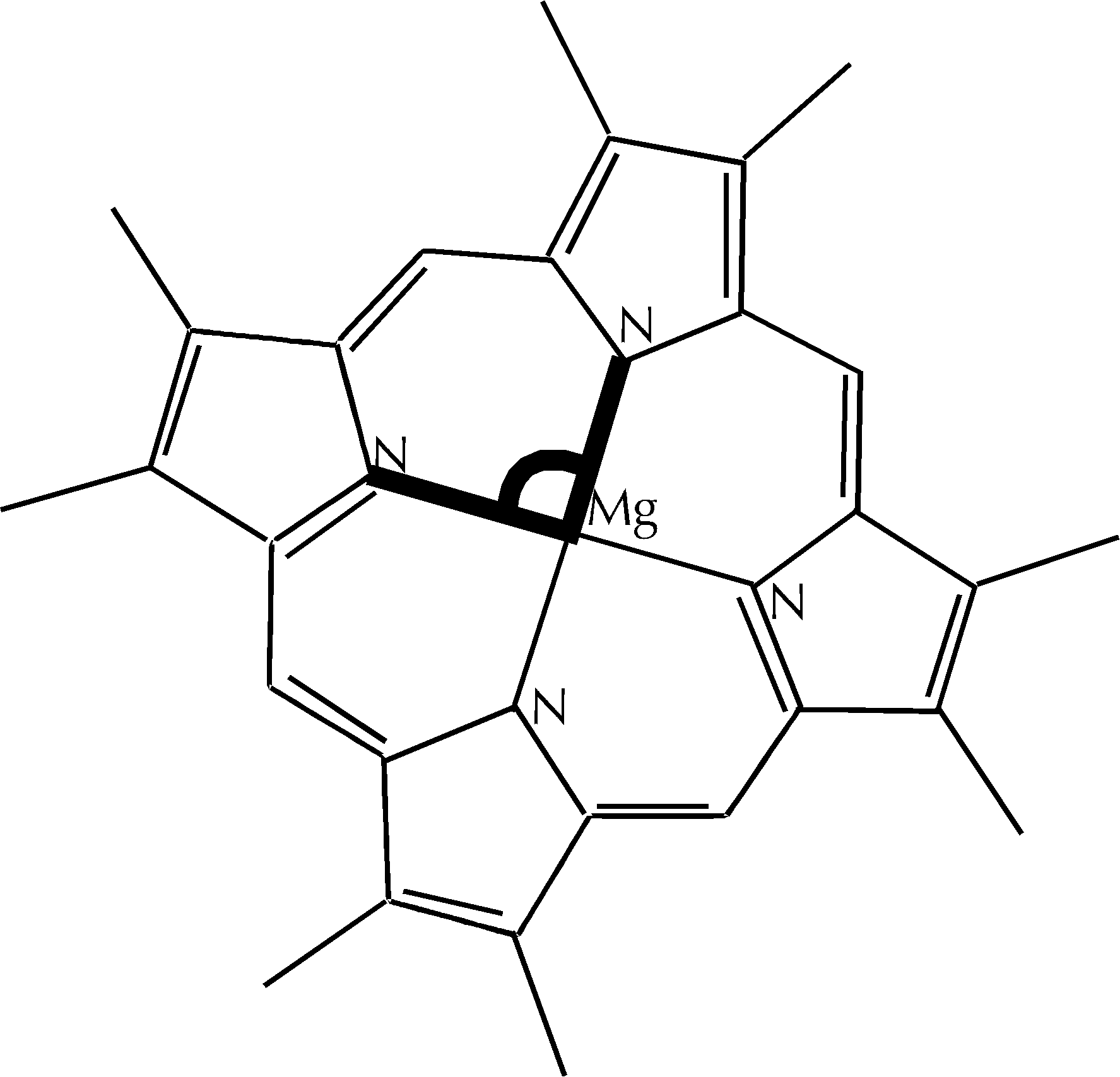 720 720 |
|
|
|
Figura -8: Rappresentazione dell'angolo N-Mg-N
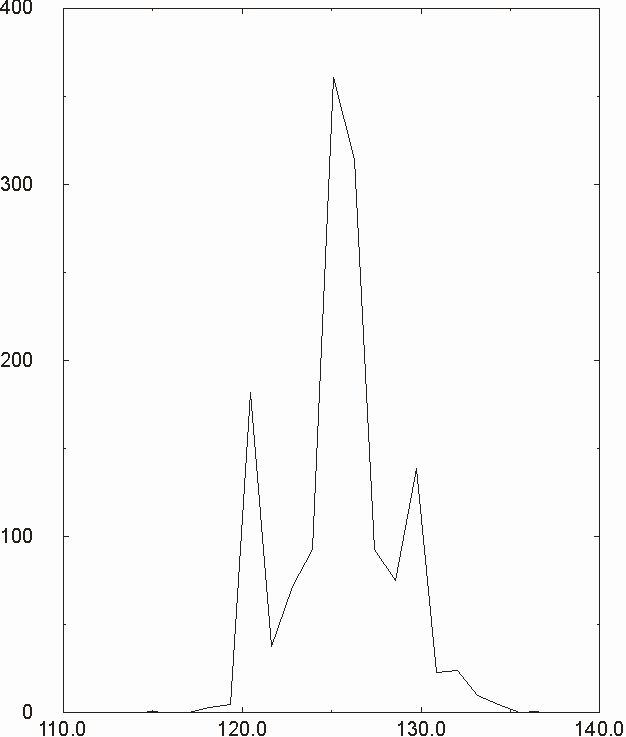 angoli Mg-N-C
angoli Mg-N-C|
|
|
|
|
|
|
|
|
|
Figura -10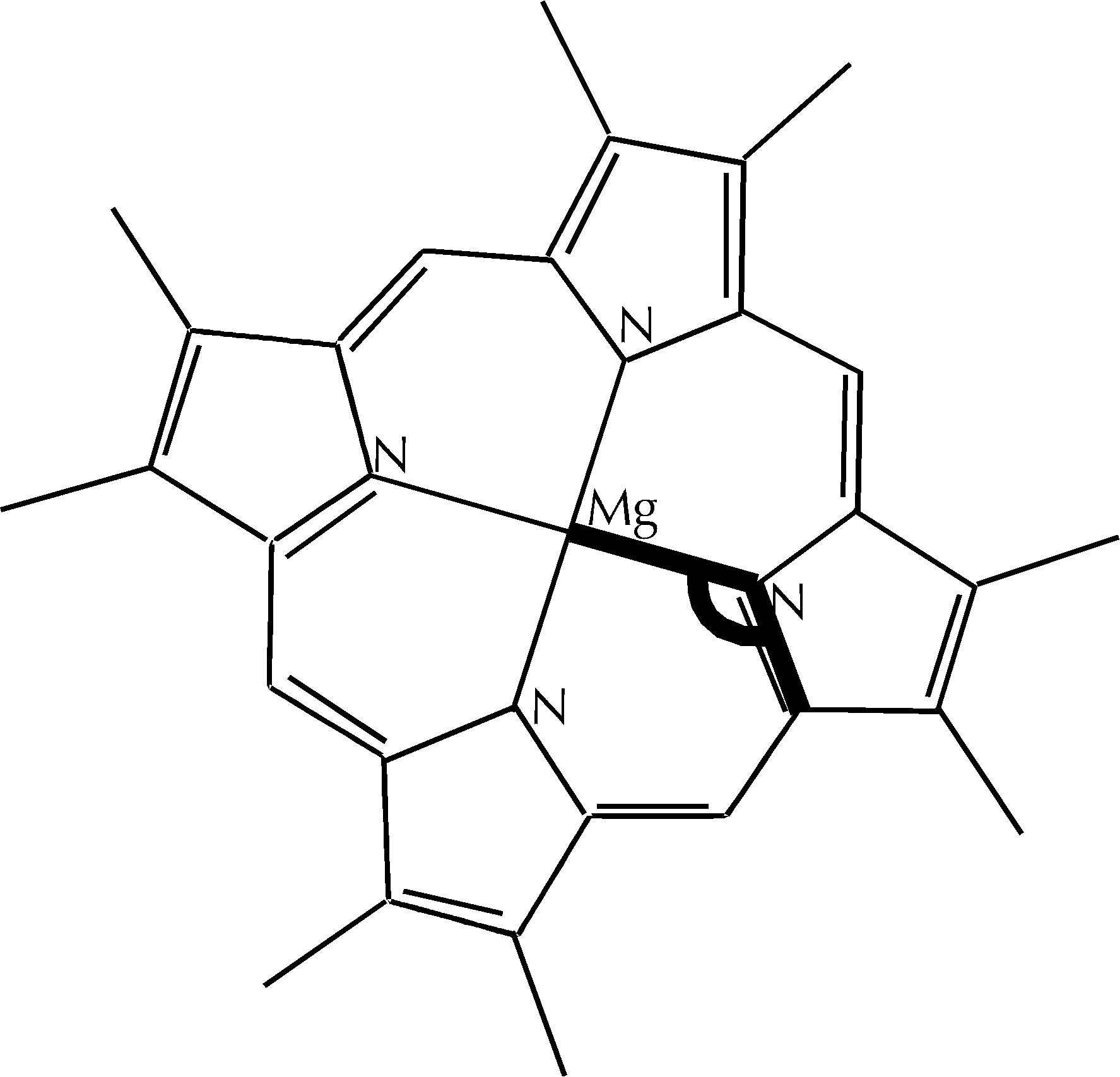 :
Rappresentazione dell'angolo Mg-N-C
:
Rappresentazione dell'angolo Mg-N-C
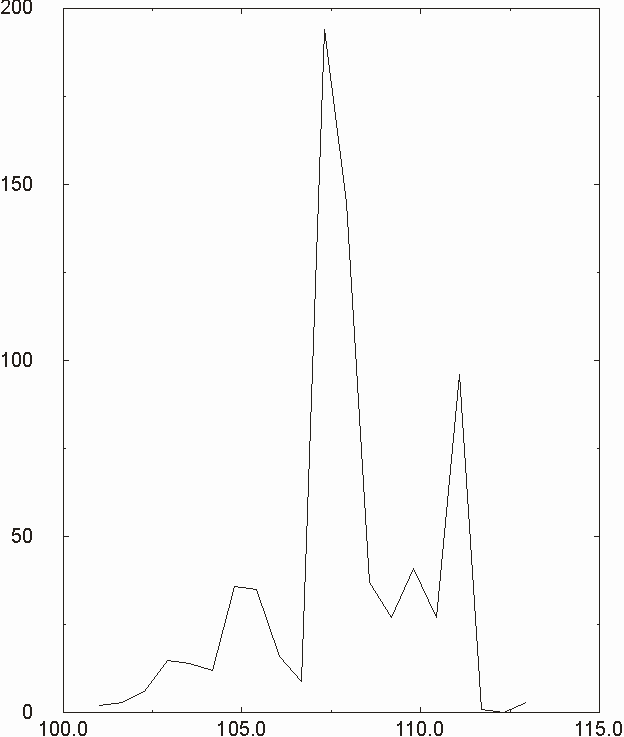 Tot Tot |
|
|
|
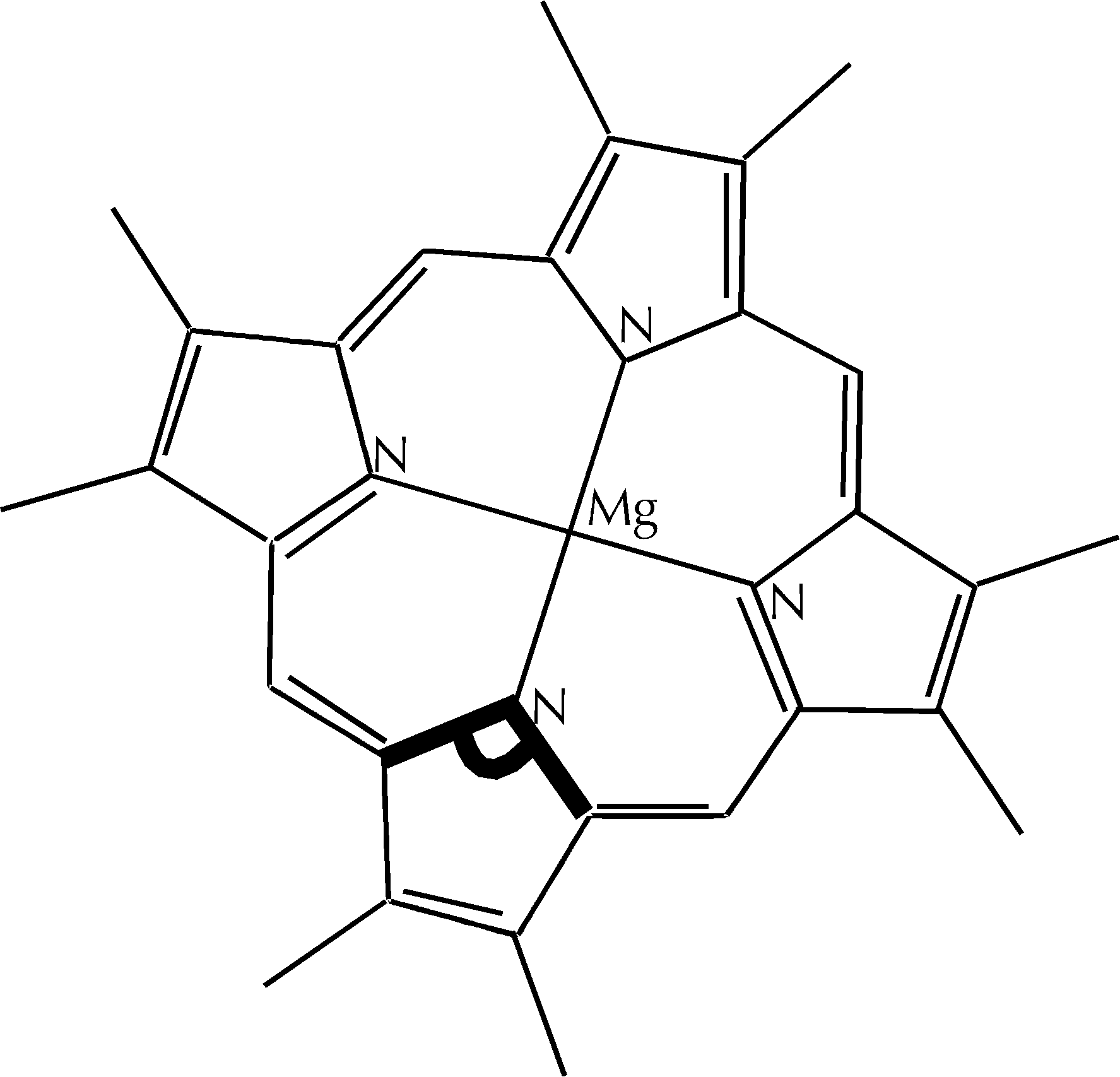 720 720 |
|
|
|
Figura -12: Rappresentazione dell'angolo C-N-C
La distribuzione dei dati è probabilmente influenzata dalle procedure utilizzate per derivare le strutture cristallografiche dai dati sperimentali. Quasi tutte le strutture sono state infatti minimizzate con il programma X-PLOR, quindi esse possono risentire del campo di forze ad esse applicato. Inoltre le strutture cristallografiche possono essere state minimizzate utilizzando diversi parametri e quindi mostrare clorofille con dati diversi da struttura a struttura. Purtroppo in nessun caso sono riportati dettagli sulla parametrizzazione del Magnesio.
L?approccio statistico utilizzato che ha permesso di tenere in considerazione tutte le strutture contenenti clorofilla finora disponibili dovrebbe in parte liberarci da queste influenze.
Per quanto riguarda alcune distribuzioni apparentemente bimodali, questo comportamento è dato dalla presenza del quinto anello all?interno della molecola di clorofilla, che causa una distorsione del macrociclo e quindi una distribuzione di angoli e di distanze che tengono conto di questa distorsione. Ovvero la distorsione provocata dal quinto anello si evidenzia nella distribuzione delle misure. Ad esempio nel caso degli angoli quasi ortogonali N-Mg-N si nota un picco sugli 86° dovuto all'angolo NC-MG-ND (nomenclatura PDB; NC appartiene all'anello II, ND all'anello III).
Per evitare l'influenza della minimizzazione, che falserebbe i dati che ci proponiamo di estrarre, sono stati considerati solo i dati relativi alle clorofille contenute nella struttura cristallografica "4bcl" (bacteriochlorophyll protein di Prosthecochloris aestuarii [Tronrud et al. 1986], cfr. § B.II.5.1.4) per i seguenti motivi:
Il concetto di potenziale di forza media è molto usato in meccanica statistica e permette di semplificare considerevolmente problemi che dipendono da un grande numero di coordinate. In pratica dato un sistema descritto da un insieme di coordinate (x,x1,...,xn) possiamo considerare la distribuzione dei valori che assume x su un insieme statistico rappresentativo del sistema.
La distribuzione di x - che chiameremo f(x) - dipende solo in maniera implicita dagli altri gradi di libertà del sistema. Ad f(x) può essere associato un potenziale (detto di forza media) secondo la relazione:
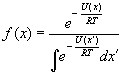
In altri termini si considera f(x) come se fosse una distribuzione di
Boltzmann corrispondente al potenziale U(x). È possibile dimostrare
che ![]() è la forza media
lungo la coordinata x [Hill 1960].
è la forza media
lungo la coordinata x [Hill 1960].
È stato assunto che, al di là delle possibili influenze dovute agli algoritmi usati per ricavare le strutture, l'insieme delle clorofille isolate dalle strutture potesse considerarsi un insieme statistico per la clorofilla e che U(x) fosse rappresentato da una funzione armonica del tipo K(x-x0)2 per ogni variabile scelta (lunghezze di legame e angoli di legame), con i parametri da determinare K e x0 che hanno rispettivamente il significato di costante di forza di richiamo e valore ottimale per la variabile in questione.
Si è quindi cercata la distribuzione Gaussiana che meglio riproducesse la distribuzione osservata e se ne è preso il valor medio come x0 e dalla larghezza a metà altezza si è ricavato K.
Le curve Gaussiane ottenute dai dati estratti da "4bcl" sono riportate nelle figure seguenti, sovrapposte agli istogrammi relativi a tutte le clorofille delle strutture cristallografiche di cui sopra, per confronto:
Figura -14: Istogrammi relativi a tutte le clorofille con sovrapposte le Gaussiane approssimanti
(vedi testo):
a: distanza Mg-N in Å
b: angolo N-Mg-N in gradi
c: angolo N-Mg-N in gradi (nella pagina seguente)
d: angolo Mg-N-C in gradi (nella pagina seguente)
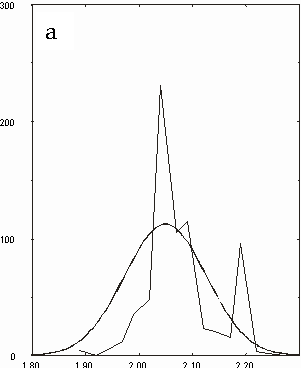 e:
angolo N-C-N in gradi (nella pagina seguente)
e:
angolo N-C-N in gradi (nella pagina seguente)
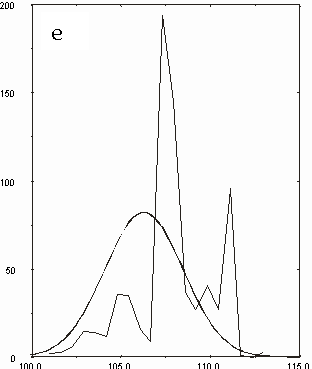
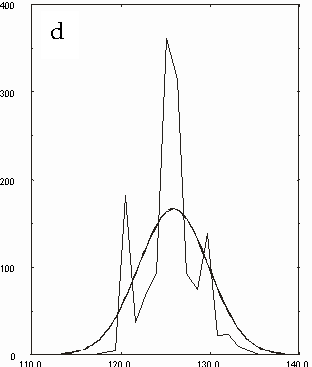
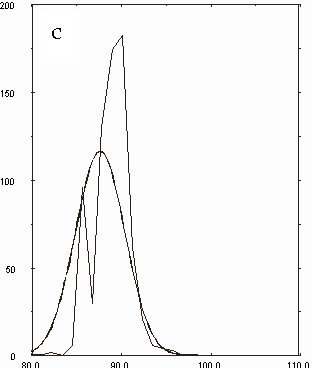 In
appendice (cfr. § F.I) sono riportate tutte le aggiunte al campo di
forza CVFF (nella fattispecie al file cvff.frc utilizzato dai programmi
Insight e Discover) qui eseguite.
Dopo aver completato il campo di forze con i dati relativi al Magnesio
presente nelle clorofille, ricostruiti i pigmenti e sistemate cariche parziali
e tipi atomici, il programma Discover è stato usato per tentare
alcune minimizzazioni energetiche.
In
appendice (cfr. § F.I) sono riportate tutte le aggiunte al campo di
forza CVFF (nella fattispecie al file cvff.frc utilizzato dai programmi
Insight e Discover) qui eseguite.
Dopo aver completato il campo di forze con i dati relativi al Magnesio
presente nelle clorofille, ricostruiti i pigmenti e sistemate cariche parziali
e tipi atomici, il programma Discover è stato usato per tentare
alcune minimizzazioni energetiche.
Il grosso problema a questo punto è quello di avere tra le mani una struttura incompleta, mancante dei "loop" e soprattutto della posizione delle lunghe (20 atomi di C) catene fitoliche.
La struttura appare anche ad un occhio inesperto poco compatta.
In effetti simulando una dinamica molecolare si evidenzia un collasso della struttura. Specialmente le clorofille vengono ad essere pesantemente influenzate e modificano la loro posizione, a volte mostrando fenomeni di "impaccamento", posizionandosi in modo da avere i piani dei macrocicli paralleli gli uni agli altri, migliorando quindi le interazioni di van der Waals.
Per evitare questo fenomeno di collasso si è provveduto ad imporre dei vincoli (in questo caso dei "constraint" sulla posizione: cfr. § B.VI.4) che conservino i dati cristallografici in nostro possesso (che sono - lo ricordiamo - solamente le posizioni dei carboni alfa delle eliche e la posizione dei cromofori - ma non il loro orientamento) e lascino libertà al resto della struttura per consentire una minimizzazione energetica e quindi un posizionamento teoricamente migliore delle catene laterali.
Gli algoritmi automatici usati per la ricostruzione delle catene laterali non riconoscono la presenza dei pigmenti nella struttura quindi, anche se la loro assegnazione è plausibile perché basata su librerie di rotameri e banche di dati strutturali, alcune catene laterali risultano posizionate molto sfavorevolmente, con forte ingombro sterico causato dalle clorofille o luteine ad esse vicine.
Le minimizzazioni energetiche (meccanica molecolare) non riescono a correggere completamente questi problemi. Causano invece distorsione di alcuni residui aminoacidici a causa della loro posizione sfavorevole (vedi Figura D-20).
La meccanica molecolare in questo caso è poco efficace perché non può modificare sostanzialmente una struttura. Non esplora lo spazio conformazionale.
Per portare questi residui in posizioni termodinamicamente più favorevoli si sono inizialmente tentate delle semplici simulazioni di dinamica molecolare di "simulated annealing" (questo termine deriva dalla procedura di annealing, letteralmente "temperamento", utilizzata per far sì che un solido raggiunga il suo stato di minima energia in seguito a riscaldamento ad alte temperature e lento raffreddamento).
Queste consistono nella simulazione di un riscaldamento (fino ad una temperatura di 1000 K) della struttura, fornendo un'energia tale da muovere le catene laterali (mediante rotazioni dei legami) e superare alcune barriere energetiche, seguito da un progressivo raffreddamento (temperatura abbassata di 100 K ogni 2 picosecondi di dinamica). Questo aiuta a riarrangiare la posizione di catene laterali (o di intere parti della struttura se questa non fosse nel caso specifico vincolata) in conformazioni teoricamente migliori.
Purtroppo questo tipo di simulazione non si è rivelato sufficiente perché alcune barriere energetiche non possono comunque essere superate dalle catene laterali, soprattutto se il superamento delle barriere implica una compenetrazione di sfere di van der Waals. Le catene laterali non possono muoversi in direzioni che comportino un eccessivo avvicinamento tra due atomi, limitando di fatto l'esplorazione completa dello spazio conformazionale per alcune situazioni strutturali. Gli impedimenti sterici bloccano quindi alcuni residui in una posizione ancora sfavorevole dal punto di vista energetico.
Il caso più evidente di questo è rappresentato dal residuo His212, bloccato dalla clorofilla A3 mentre la sua posizione dovrebbe essere tale da coordinare B3 (cfr. § B.II.6.1.3 e Figura B-9), come evidenziato in Figura D-20 (in alto).
Per superare tali problemi e raggiungere il minimo globale dell'energia potenziale relativa alla struttura in esame è stato sfruttata una procedura di simulated annealing in cui tutte le costanti delle forze interagenti nella simulazione sono inizialmente poste a livelli praticamente nulli e successivamente incrementate gradualmente secondo andamenti lineari, geometrici od esponenziali. Questo tipo di protocollo fu proposto inizialmente da Nilges, Clore e Gronenborn [1988] per la determinazione strutturale a partire da vincoli ottenuti da misure NMR.
In questo modo all?inizio della simulazione gli atomi sono liberi di muoversi indipendentemente dalla loro posizione (e intorno chimico) iniziale creando una disposizione casuale che poi viene gradualmente ristretta e portata (incrementando il peso delle forze in gioco) alla configurazione fisicamente corretta.
Il protocollo è stato opportunamente modificato per applicarlo al nostro sistema. Vari problemi si sono posti e sono stati affrontati e risolti:
La struttura iniziale viene generata in maniera casuale, assegnando
valori aleatori alle coordinate x y e z di ogni atomo del sistema ma mantenendo
la topologia dei legami.
Le costanti di forza per tutte le componenti energetiche di legame (costanti di lunghezza di legame, d'angolo) vengono poste a 1/100000 del valore per esse tabulato nel campo di forze. Le costanti di forza per la componente di van der Waals sono poste ad 1/10000 del loro valore tabulato.
La dinamica viene inizializzata ovvero si simula una temperatura di 1000 K mediante un'assegnazione di vettori velocità agli atomi del sistema secondo una distribuzione di Maxwell-Boltzmann.
La prima fase della simulazione consiste in cinquanta cicli di dinamica molecolare, ognuno della durata di un picosecondo (ps), con un timestep di 1 fs ed una temperatura di 1000 K.
All'inizio di ciascun ciclo le costanti di forza (escluse quelle di van der Waals) sono aumentate moltiplicandole per 1.25, scalandole così in maniera esponenziale fino a far loro riassumere il valore originario tabulato al quarantesimo picosecondo.
Le costanti di repulsione atomica di van der Waals iniziano a crescere al trentesimo ciclo, moltiplicate ad ogni passo di un fattore 1.5, fino a raggiungere il loro valore originario al picosecondo 50.
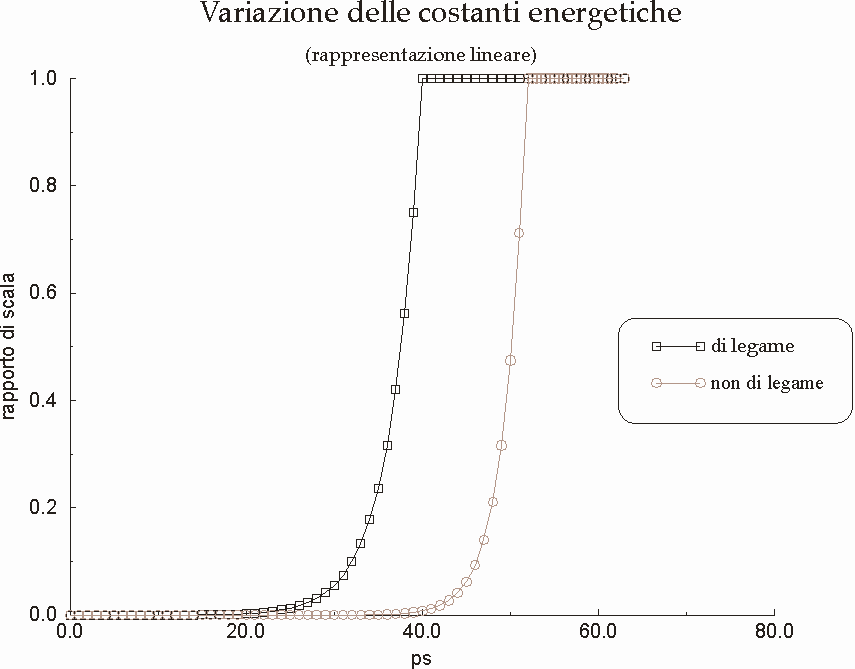
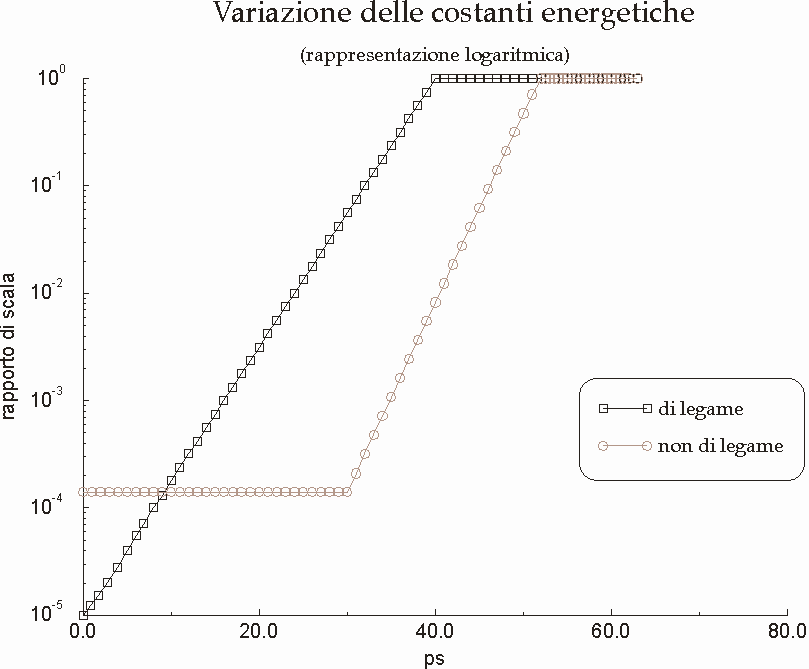 Il
valore delle costanti repulsive è molto basso durante questi primi
stadi, quindi permette agli atomi di avvicinarsi notevolmente, abbassando
così le barriere energetiche da superare per migliorare localmente
la struttura.
Il
valore delle costanti repulsive è molto basso durante questi primi
stadi, quindi permette agli atomi di avvicinarsi notevolmente, abbassando
così le barriere energetiche da superare per migliorare localmente
la struttura.
Figura -15: Variazione delle costanti energetiche nel corso della simulazione
La seconda fase del protocollo consiste in 20 cicli da 1 ps l'uno in cui le costanti di forza sono mantenute al loro valore originario e la temperatura viene gradualmente e linearmente raffreddata fino a raggiungere i 300 K, ovvero la temperatura ambiente.
Seguono 2 picosecondi di dinamica a 200 K come pre-minimizzazione e quindi 8000 cicli di minimizzazione energetica.
Per orientare la simulazione in modo tale che la struttura finale fosse compatibile con le informazioni cristallografiche si è provveduto ad imporre dei vincoli strutturali (cfr. § B.VI.4). Con una costante di forza indipendente (denominata FRMS) gli atomi Ca (i carboni alfa dello scheletro polipeptidico), i quattro atomi di azoto del macrociclo della clorofilla e i carboni delle luteine vengono spinti a riassumere le posizioni della struttura cristallografica. È questa la componente che ricrea la struttura tridimensionale (le eliche transmembrana e la disposizione dei pigmenti) a partire dalla randomizzazione iniziale.
FRMS cresce esponenzialmente insieme con le costanti di legame da un valore quasi nullo (0.0002 kcal/(mol× Å2)) fino ad un massimo di 20 kcal/(mol× Å2) in corrispondenza del quarantesimo picosecondo e poi viene mantenuta costante su quel valore. Questo permette il libero movimento degli atomi all'inizio della simulazione per poi gradualmente spingere la stessa verso la configurazione tridimensionale riproducente le eliche transmembrana come da struttura cristallografica.
Per mantenere la corretta chiralità dei residui aminoacidici, che verrebbe persa in seguito alla disposizione casuale iniziale degli atomi e al rilassamento delle costanti energetiche, è stato aggiunto un nuovo termine di forza al sistema, controllato da una costante indipendente denominata FKCHIR.
Dopo vari tentativi empirici si è deciso di applicare tale costante in modo molto rilevante all'inizio della simulazione per poi calarne gradualmente l'influenza.
FKCHIR all'inizio viene impostata a 300 kcal/(mol× Å2) e decresciuta esponenzialmente fino ad un valore costante di 50 kcal/(mol× Å2) al cinquantesimo picosecondo.
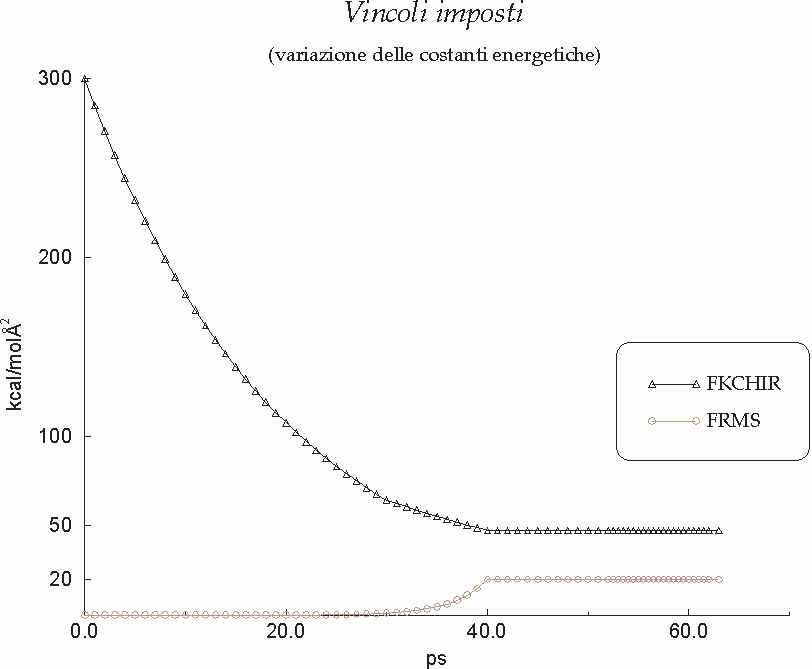 In figura l'andamento delle
due costanti suddette:
In figura l'andamento delle
due costanti suddette:
Figura -16: Variazione delle costanti energetiche per i vincoli imposti nel corso della simulazione
Il timestep prescelto (1 fs) è quello normalmente adoperato nelle dinamiche molecolari ma ha presentato parecchi problemi nella dinamica a costanti di forza rilassate. La ragione è stata individuata nella difficoltà di mantenere la simulazione alla temperatura impostata a causa dell'alta mobilità raggiunta dagli atomi di idrogeno. Una soluzione di questo verrebbe dalla modificazione del timestep, ad esempio con l'uso di un timestep di 0.1 fs. Questo sarebbe però computazionalmente eccessivo richiedendo 10 volte il numero di calcoli e quindi accrescendo di dieci volte la durata della simulazione (dalle 12 ore circa alle 120 ore).
La soluzione qui adottata è di accrescere a 10 u.m.a. (unità di massa atomica) la massa degli atomi di idrogeno (la cui massa è circa 1 u.m.a.). In questo modo essi vengono frenati, possedendo un'inerzia 10 volte maggiore, senza altre influenze sul loro comportamento e su quello dell'intero sistema [Pomès e McCammon 1990].
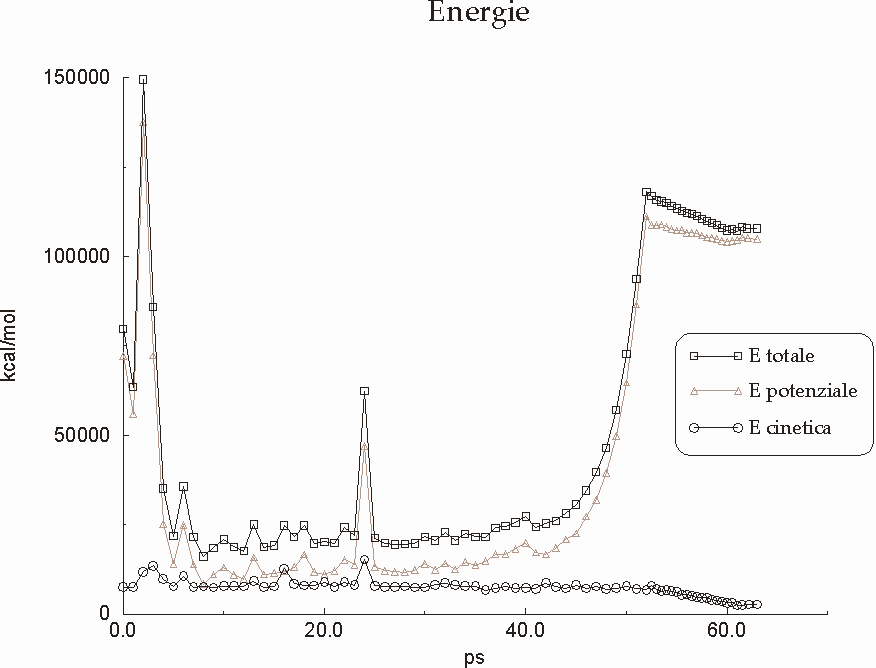 Di seguito i grafici riportanti
l'andamento delle energie (potenziale, cinetica e totale: la somma delle
due) e delle temperature (quella impostata dal protocollo e quella invece
riscontrata nella simulazione) per la proteina LHC II sottoposta al simulated
annealing così impostato:
Di seguito i grafici riportanti
l'andamento delle energie (potenziale, cinetica e totale: la somma delle
due) e delle temperature (quella impostata dal protocollo e quella invece
riscontrata nella simulazione) per la proteina LHC II sottoposta al simulated
annealing così impostato:
Figura -17: Grafico rappresentante l'andamento delle energie durante il simulated annealing
Figura -18: Grafico rappresentante l'andamento delle temperature durante il simulated annealing
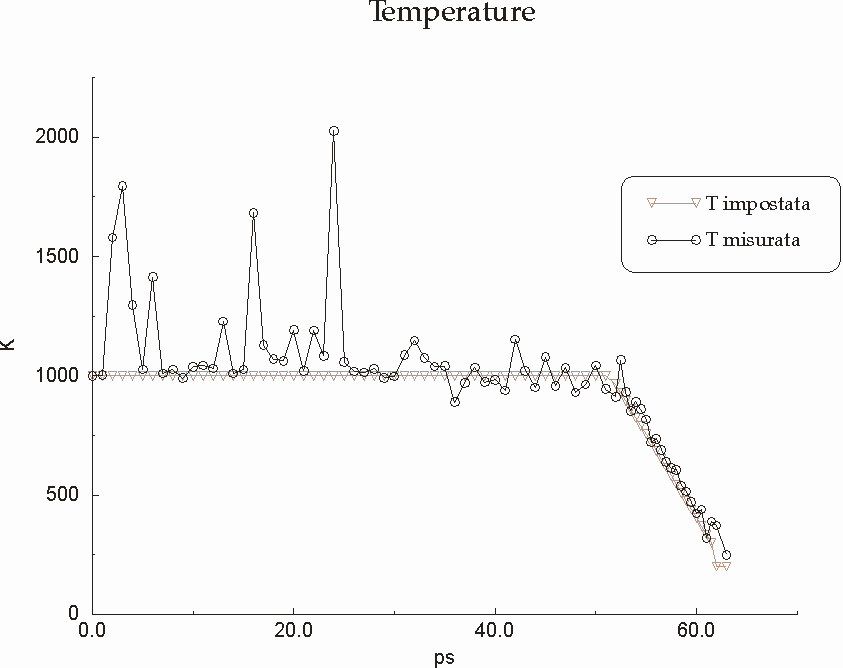
L'energia potenziale presenta una rapida discesa dopo l'alto picco iniziale (generato dalla sovrapposizione di alcuni atomi in seguito alla randomizzazione) dovuta alla distensione della struttura e alle bassissime costanti di forza.
Quando queste diventano rilevanti, soprattutto quelle di repulsione elettrostatica di van der Waals, si assiste ad un esponenziale innalzarsi di questa energia, seguita dall'abbassarsi dell'energia cinetica dovuta al raffreddamento della fase 2.
I picchi nell'energia potenziale che si osservano durante la fase 1 sono probabilmente dovuti all'intervento a volte brusco della componente che si occupa del mantenimento della chiralità che deve a volte invertire rispetto ad un piano intere parti della struttura per ricostruire la corretta orientazione chirale.
L'azione di questa componente è rilevabile anche nei seguenti
fotogrammi che illustrano graficamente l'andamento della simulazione nel
tempo:
Nella pagina seguente:
Il fotogramma corrispondente al ps 37 mostra eliche e pigmenti riformati, nessun brusco riarrangiamento chirale mentre le catene laterali sono ancora in movimento e alcuni anelli aminoacidici rimangono non planari.
Infine la struttura è riformata completamente senza distorsioni energetiche, con le catene laterali portate ad assumere una configurazione più corretta relativa all?intorno costituito dagli altri residui e dai pigmenti, abbassando notevolmente l?energia totale del sistema calcolata da questo software.
Le misure di energie visibili nei grafici e nel seguente schema danno un'indicazione relativa della distanza del sistema da uno stato di equilibrio e non possono essere interpretate come misure assolute di energia, dipendendo questi valori dai vincoli imposti e dalle costanti del campo di forze.
La minimizzazione energetica condotta al termine della dinamica registra
questa situazione energetica finale, per mole:
| 2160.771 kcal energia
totale
450.399 kcal energia relativa alle distanze di legame 1015.555 kcal energia relativa agli angoli di legame 270.593 kcal energia relativa agli angoli di torsione 71.063 kcal energia relativa alle interazioni non planari -267.821 kcal energia relativa alle interazioni Coulombiane 620.982 kcal energia relativa alle interazioni di non legame di cui: 4697.090 kcal componente repulsiva
-4076.108 kcal componente attrattiva
|
Per confronto la simulazione di simulated annealing senza rilassamento delle costanti di forza mostrava un'energia totale finale di 2838 kcal/mol.
Il risultato finale è una struttura in cui le catene laterali ricostruite hanno ora una posizione plausibile, determinata dalle interazioni fisiche e chimiche con il loro intorno molecolare, come esemplificato graficamente in Figura D-20.
Analizzando la struttura si riscontrano comunque alcune distorsioni non risolte, ad esempio a carico della planarità dei residui His 68 e Trp 128 o relativamente alle distanze di legame e agli angoli di legame.
Questo è causato dal constraint strutturale imposto sulla posizione dei Ca e su quella degli atomi costituenti i pigmenti che sono forzati a mantenere la posizione originale anche nelle minimizzazioni successive al simulated annealing. Considerando che la risoluzione della struttura cristallografica di partenza è di 3.4 Å, è possibile che vi sia incertezza nelle coordinate. Peraltro la struttura non è mai stata depositata in PDB e quindi le coordinate su cui si è lavorato in questa tesi vanno senz'altro intese come non raffinate. A riprova di ciò abbiamo considerato le distanze fra Ca consecutivi che risultano variare da 3.67 Å a 3.95 Å con un valore medio di 3.805 ± 0.049 Å. In un sottoinsieme di strutture ad alta risoluzione depositate in PDB tale distanza risulta essere praticamente fissata a 3.78 Å ± 0.05 Å [Fogolari et al. 1996].
L'orientazione del macrociclo delle clorofille potrebbe essere affetta da una simile imprecisione che potrebbe portare ai contatti sfavorevoli osservati.
Essendo però la posizione dei Ca e dei pigmenti l'unica informazione strutturale disponibile, si è preferito mantenere queste invariate ed accettare le corrispondenti distorsioni derivanti. La struttura ricostruita offre comunque la possibilità, al di là di queste distorsioni, di studiare la plausibilità delle interazioni tra i residui e fra residui e clorofille e quindi di formulare ipotesi di modello.
La simulazione ha infatti portato alla luce nuove ipotesi di modello per alcune interazioni chiave tra gli aminoacidi di cui si discuterà in seguito (cfr. § D.VIII).
Una minimizzazione in cui venga rimosso il constraint ad atomi fissati sui Ca , sostituendolo con un restraint di tipo tethering (cfr. § B.VI.4), e in cui venga forzata la planarità delle unità peptidiche, riduce grandemente tali distorsioni modificando leggermente la posizione originaria di questi, come riportato in appendice (cfr. § F.IV). La costante di forza usata nel tethering è pari a 32 kcal/(mol× Å2) per ogni Ca , con 24 kcal/(mol× rad2) di costante di forza per il vincolo sull'angolo diedro w , che definisce la planarità delle unità peptidiche.
Le posizioni e conformazioni delle catene laterali relative alle ipotesi
di modello che vengono in seguito formulate non sono sostanzialmente modificate
da tale minimizzazione.
Nella pagina seguente:
di simulated annealing a costanti di forza rilassate
In alto: residuo Istidina 212 dopo minimizzazione o normale simulated annealing. L'aminoacido si trova in posizione sfavorevole non potendo oltrepassare la clorofilla a3 (a destra in figura) per arrivare a coordinare la clorofilla b3 (a sinistra) e questo causa una deformazione della sua usuale conformazione (con anello planare).
In basso: lo stesso residuo in seguito ad applicazione del protocollo a costanti di forza rilassate. L'aminoacido ora si trova in posizione favorevole per la coordinazione e non presenta distorsione nella conformazione.
Un allineamento tra il complesso maggiore LHC II e le antenne minori
relativo alle proteine del mais è pubblicato in letteratura ([Bassi
et al. 1997]).
Si è qui deciso di allineare in modo indipendente la proteina LHC II di pisello alle antenne minori del mais al fine di creare un allineamento guida per poi allineare tutte le proteine della famiglia Lhcb di cui la sequenza è nota e disponibile in banca dati.
La qualità dell'allineamento tra le sequenze omologhe determina infatti la correttezza dei modelli ricostruiti per omologia (cfr. § B.VI), uno degli scopi prefissati in questo lavoro di tesi. Un allineamento tra il maggior numero di sequenze omologhe è inoltre necessario per uno studio della covarianza aminoacidica (cfr. § B.VII) e per ricavare informazioni strutturali dall'analisi delle sequenze.
Sono stati usati i due programmi "ClustalW" (cfr. § C.II.1) e "Macaw" (cfr. § C.II.3) per un approccio parallelo di due diverse metodologie.
In particolare i due programmi presentavano un disaccordo tra l'allineamento dell'elica transmembrana C di LHC II e CP24.
Il disaccordo è stato risolto con l'uso dell'algoritmo di predizione eliche transmembrana (cfr. § B.V.3.2) disponibile nel server PredictProtein (cfr. § C.II.4).
L'allineamento dato da ClustalW per le eliche CLHC II-CCP24:
LHC II SILAIWATQVILMG-AVEG---Y-RIA
CP24 VESKRWVDFFNPDSQAVEWATPWSRTA
L'allineamento CLHC II-CCP24 di Macaw:
LHC II SILAIWATQVILMGAVEGYRIA
CP24 SFGSLLGTQLLLMGWVESKRWV
PredictProtein predice la posizione per l'elica transmembrana C di CP 24 nel seguente modo (gli aminoacidi marcati in grassetto sono quelli aventi maggiore probabilità di presenza in elica transmembrana):
CP24 ADPTRIAPFSFGSLLGTQLLLMGWVESKR
Poiché vogliamo allineare le eliche transmembrana C viene preferito l'allineamento di Macaw.
Figura -21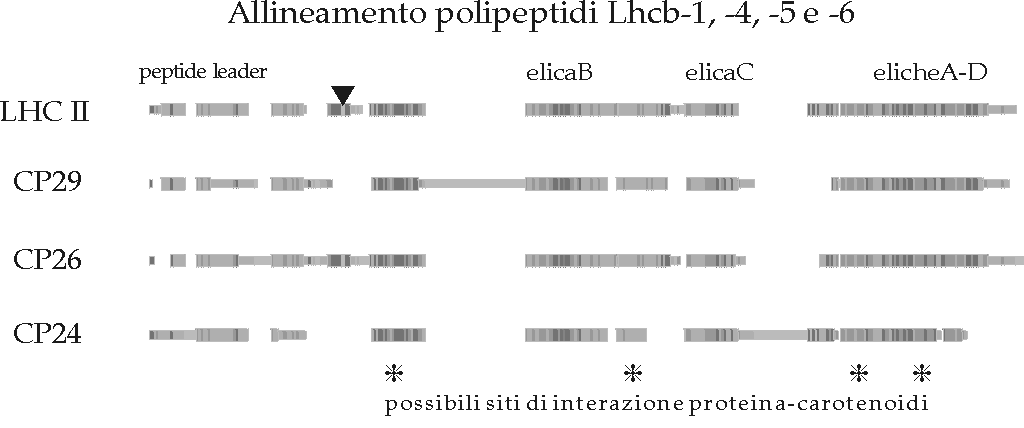 :
Rappresentazione a blocchi di omologia dell'allineamento risultante
:
Rappresentazione a blocchi di omologia dell'allineamento risultante
(colorazione per analogie aminoacidiche; la posizione del triangolo indica un sito coinvolto nella trimerizzazione di LHC II, conservato anche in CP 26: WYGPDRVKYL [Hobe et al. 1995], aminoacidi 16-25 in CB22_PEA)
L'allineamento risultante è il seguente (sottolineati i possibili siti di interazione proteina-carotenoidi ipoteticamente derivanti dalla sequenza consenso WFDPL [Pichersky e Jansson 1996]; marcati in grassetto alcuni aminoacidi chiave importanti per la trattazione, cfr. anche § D.VI.1):
LHC II 1 RKSATTKKVASSGSP---------WYGPDRVKYLGPFSG---E
CP_29 1 RFGFGLGGKAKPAPKKVAKTSTSSDRPL----WFPGAV-------------
CP_26 1 LFSKKPAQKPKPSAVSSSSPDISDELAKWYGPDRRIYLPDGLLDRSE
CP_24 1 AAAAAKKS---------------------WIPAIKSDAEIV-------
LHC II 32 SPSYLTGEFPGDYGWDTAGLSAD---------
CP 29 35 APDYLDGSLVGDYGFDPFGLGKPVEYLQFELD
CP 26 48 VPEYLTGEVPGDYGYDPFGLGKK---------
CP 24 21 NPPWLDGSLPGDFGFDPLGLGKD---------
LHC II ----------------------------------
CP 29 67 SLDQNLAKNEAGGIIGTRFESSEVKSTPLQPYSE
CP 26 ----------------------------------
CP 24 ----------------------------------
ELICA B
LHC II 55 PETFSKNRELEVIHSRWAMLGALGCVFPELLSRNG
CP 29 101 VFGLQRFRECELIHGRWAMLATLGALSVEWLTGVT
CP 26 71 PEDFAKYQAYELIHARWAMLGAAGAVIPEACNKFG
CP 24 44 PAFLKWYREADVIHGRWAMAAVLGIFVGQAWSGIP
LHC II 90 VKFG-EAVWFKAGSQIFSEGGLDYLGNPSLVHAQ
CP 29 136 --------WQDAGKVELVDGSS-YLGQPLPF---
CP 26 106 ANCGPEAVWFKTGALLLDGNTLSYFGNSIPI---
CP 24 79 --------WFEAGADPTRIAPF------------
ELICA C
LHC II 123 SILAIWATQVILMGAVEGYRIA---------------
CP 29 158 SISTLIWIEVLVIGYIEFQRNAELDPEKR--------
CP 26 137 NLVVAVIAEVVLVGGAEYYRIINGL------------
CP 24 93 SFGSLLGTQLLLMGWVESKRWVDFFNPDSQAVEWATP
LHC II -----GGPLGEVVDPLYPGG-SFDPLGLAD----------
CP 29 ---------------LYPGGSYFDPLGLAA----------
CP 26 ----------DLEDKLHPGG-PFDPLGLAS----------
CP 24 WSRTAENFANFTGEQGYPGGKFFDPLAL-AGTSRDGVYIP
ELICA A
LHC II 169 DPEAFAELKVKELKNGRLAMFSMFGFFVQAI
CP 29 202 DPEKKERLQLAEIKHARLAMVAFLGFAVQAA
CP 26 181 DPDQAAILKVKEIKNGRLAMFSMFAFFIQAY
CP 24 169 DVDKLERLKLAEIKHARIAMLAMLAFYFEAG
ELICA D
LHC II 200 VTGKGPLENLADHLADPVNNNA
CP 29 233 ATGKGPLNNWATHLSDPLHTTI
CP 26 212 VTGEGPVENLAKHLSDPFGNNL
CP 24 200 Q-GKTRLGALGL----------
L'allineamento verificato è stato esteso a tutte le sequenze
conosciute di proteine antenna appartenenti alla famiglia multigenica dei
geni Lhcb (cfr. § D.I) con l'uso della funzione "profile alignment"
di ClustalW che usa un allineamento guida per allineare nuove sequenze.
Sono state inoltre inserite le informazioni relative alla posizione delle
eliche transmembrana nell'allineamento guida in modo da influenzare selettivamente
la possibilità di apertura di gap. La penalità per
l'apertura di questi (cfr. § B.V.2) è molto maggiore nelle
zone indicate aventi struttura secondaria ordinata, favorendo il mantenimento
compatto di queste: sono state utilizzate penalità di 7 per le zone
dei residui appartenenti alle eliche transmembrana, 5 per il corto loop
tra le eliche A e D, 2 per la zona conservata nel loop all'N-terminale
(32-54 in LHC II di pisello).
Si noti che nel nostro caso l'albero filogenetico vuole solo fornire una rappresentazione grafica della distanza di omologia di sequenza tra le diverse proteine e non costituisce un'analisi filogenetica che richiederebbe studi più approfonditi, soprattutto a livello di DNA.
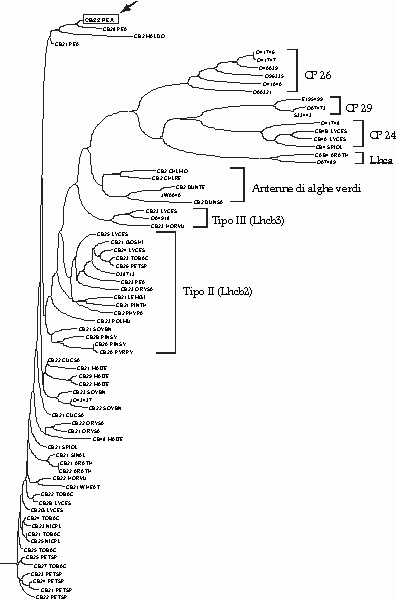 La distanza (indicata dalla
lunghezza orizzontale dei rami intercorrenti fra due sequenze) tra le antenne
minori e le LHC II di tipo I è rilevante, ma LHC II di pisello (indicata
con una freccia nell'albero) appare tra le tipo I più simili alle
antenne minori. Questo è evidenziabile dalla vicinanza in termini
di "numero di nodi". Ovvero vi sono molte meno diramazioni (i nodi dell'albero)
intercorrenti tra essa e le antenne minori di quante ce ne siano tra queste
e quasi tutte le altre LHC II di tipo I.
La distanza (indicata dalla
lunghezza orizzontale dei rami intercorrenti fra due sequenze) tra le antenne
minori e le LHC II di tipo I è rilevante, ma LHC II di pisello (indicata
con una freccia nell'albero) appare tra le tipo I più simili alle
antenne minori. Questo è evidenziabile dalla vicinanza in termini
di "numero di nodi". Ovvero vi sono molte meno diramazioni (i nodi dell'albero)
intercorrenti tra essa e le antenne minori di quante ce ne siano tra queste
e quasi tutte le altre LHC II di tipo I.
Figura -22: Rappresentazione ad albero della distanza tra le sequenze proteiche nella famiglia Lhcb
L'albero evidenzia inoltre come le antenne minori abbiano omologia con (in ordine decrescente di omologia) le antenne di alghe verdi, le tipo III e le tipo II.
Due proteine appartenenti al complesso antenna del fotosistema I (LHC I, geni Lhca) sono riportate nell'albero per indicare il loro rapporto con la famiglia Lhcb: esse sono più vicine in omologia alle antenne minori (in particolar modo a CP 24 e CP 29) di quanto non siano al complesso LHC II.
CP 24 presenta invece un'inserzione di circa 35 aminoacidi tra il secondo ed il terzo blocco (ovvero a monte del loop conservato che precede l'elica A), nonché la mancanza dell'elica D.
Segue l'allineamento tra i due blocchi derivanti dalla probabile duplicazione (dopo aver eliminato l'inserzione che in CP 29 divide il primo blocco) per le sequenze dell'allineamento completo (cfr. § D.V.1):
LHCII blocco 1 YLTGEFPGDYGWDTAGLSAD-------PETFS--KNRELEVIHSRWAMLGALGCVFPELLSRNG 89
LHCII blocco 3 VVDPLYPGG-SFDPLGLADD-------PEAFA--ELKVKELKNGRLAMFSMFGFFVQAIVTGKG 204
CP26 blocco 1 YLTGEVPGDYGYDPFGLGKK-------PEDFA--KYQAYELIHARWAMLGAAGAVIPEACNKFG 105
CP26 blocco 3 LEDKLHPGG-PFDPLGLASD-------PDQAA--ILKVKEIKNGRLAMFSMFAFFIQAYVTGEG 216
CP29 blocco 1 YLDGSLVGDYGFDPFGLGKP-------VEVFGLQRFRECELIHGRWAMLATLGALSVEWLTGVT 135
CP29 blocco 3 ----LYPGGSYFDPLGLAAD-------PEKKE--RLQLAEIKHARLAMVAFLGFAVQAAATGKG 237
CP24 blocco 1 WLDGSLPGDFGFDPLGLGKD-------PAFLK--WYREADVIHGRWAMAAVLGIFVGQAWSGIP 78
CP24 blocco 3 ----GYPGGKFFDPLALAGTSRDGVYIPDVDKLERLKLAEIKHARIAMLAMLAFYFEAGQ-GKT 203
(in grassetto aminoacidi con identità per tutte le sequenze, in grassetto corsivo analogia per tutte le sequenze)
Anche in questo tipo di analisi si può notare la maggior vicinanza genetica di CP26 a LHC II (più alto numero di residui identici), come mostrato dalla raffigurazione ad albero filogenetico (cfr. § D.V.3) dell'allineamento normale.
Usando il programma AliAna (cfr. § C.II.6) è possibile calcolare
la percentuale di conservazione per ogni residuo, ovvero un'analisi di
ricorrenza su un allineamento ed una regione aminoacidica selezionati.
Per le (51) sequenze tipo I e II e la regione disponibile nella struttura
cristallografica (100 aminoacidi costituenti le quattro eliche B C A e
D), l'analisi è la seguente (ad esempio in posizione 6 il 78% delle
sequenze esaminate porta una Lisina, il rimanente 22% un'Arginina):
| Gli aminoacidi sono indicati
con la loro abbreviazione ad una lettera (cfr. § B.I).
La numerazione della posizione (da 1 a 100) si basa sull'allineamento inserito. Zona 1-35: elica transmembrana B (aminoacidi 55-89 in CB22_PEA). Zona 36-55: elica transmembrana C (aminoacidi 123-142 in CB22_PEA). Zona 56-100: elica transmembrana A ed elica anfipatica D (aminoacidi 170-214 in CB22_PEA). |
||||||||||||||||||||
|
|
A | C | D | E | F | G | H | I | K | L | M | N | P | Q | R | S | T | V | W | Y |
|
|
1.00 | |||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.96 | 0.04 | ||||||||||||||||||
|
|
0.78 | 0.22 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.02 | 0.51 | 0.02 | 0.45 | ||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
0.02 | 0.96 | 0.02 | |||||||||||||||||
|
|
0.02 | 0.96 | 0.02 | |||||||||||||||||
|
|
0.96 | 0.04 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.04 | 0.02 | 0.02 | 0.92 | ||||||||||||||||
|
|
0.96 | 0.04 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.14 | 0.86 | ||||||||||||||||||
|
|
0.08 | 0.92 | ||||||||||||||||||
|
|
0.65 | 0.02 | 0.33 | |||||||||||||||||
|
|
0.24 | 0.75 | 0.02 | |||||||||||||||||
|
|
0.02 | 0.96 | 0.02 | |||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
0.02 | 0.63 | 0.04 | 0.25 | 0.06 | |||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.33 | 0.02 | 0.65 | |||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.80 | 0.12 | 0.08 | |||||||||||||||||
|
|
0.16 | 0.84 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
0.02 | 0.96 | 0.02 | |||||||||||||||||
|
|
0.14 | 0.84 | 0.02 | |||||||||||||||||
|
|
0.92 | 0.02 | 0.02 | 0.04 | ||||||||||||||||
|
|
0.04 | 0.94 | 0.02 | |||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
0.04 | 0.96 | ||||||||||||||||||
|
|
0.96 | 0.04 | ||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.96 | 0.02 | 0.02 | |||||||||||||||||
|
|
0.63 | 0.35 | 0.02 | |||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.98 | 0.02 | ||||||||||||||||||
|
|
0.33 | 0.67 | ||||||||||||||||||
|
|
0.02 | 0.96 | 0.02 | |||||||||||||||||
|
|
0.02 | 0.98 | ||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.76 | 0.06 | 0.02 | 0.08 | 0.02 | 0.06 | ||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
1.00 | |||||||||||||||||||
|
|
0.12 | 0.80 | 0.08 | |||||||||||||||||
|
|
0.88 | 0.02 | 0.02 | 0.02 | 0.04 | 0.02 | ||||||||||||||
Il consenso aminoacidico (conservazione di sequenza, indicando per ogni
posizione 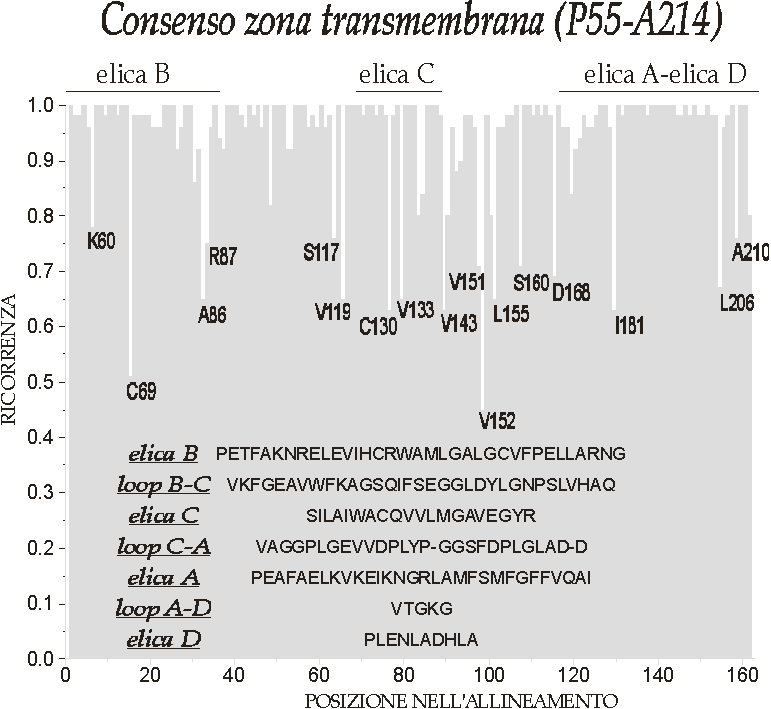 l'aminoacido
più conservato) può essere reso graficamente:
l'aminoacido
più conservato) può essere reso graficamente:
Figura -23: Ricorrenza aminoacidica e sequenza consenso per la zona transmembrana
delle proteine LHC II di tipo I e II. La numerazione (relativa a LHC II di pisello) per i segmenti indicati è la seguente: 55-89 (elica B), loop B-C (90-122), elica C (123-142), loop C-A (143-169), elica A (170-199), loop A-D (200-204), elica D (205-214). Sono indicati con la loro sigla e posizione gli aminoacidi presenti nelle posizioni con ricorrenza massima inferiore a 0.8
Passando ad uno studio per analogia invece che per identità (cfr.
§ B.IV), utilizzando una categorizzazione in AliAna ("FIV") che tenga
conto di cinque classi di aminoacidi (apolari, polari
- escluse le ammine, carichi positivamente - acidi, carichi
negativamente - basici, polari amminici, cfr.
§ C.II.6), l'analisi è la seguente (mostrata per le posizioni
presentanti divergenza):
| allin. |
|
apol | pol | pos | neg | amm | Sequenze divergenti dal consenso (indicato l'aminoacido ivi presente) |
| 2 |
|
0.02 | 0.98 | CB21_PETSP (A) | |||
| 3 |
|
0.98 | 0.02 | CB2B_PINSY (N) | |||
| 9 |
|
0.02 | 0.98 | CB21_PETSP (K) | |||
| 15 |
|
0.04 | 0.96 | CB22_HORVU (G), CB2_PHYPA (A) | |||
| 16 |
|
0.02 | 0.98 | CB22_CUCSA (T) | |||
| 17 |
|
0.98 | 0.02 | CB2_MALDO (C) | |||
| 18 |
|
0.98 | 0.02 | CB21_PETSP (T) | |||
| 20 |
|
0.98 | 0.02 | CB2_MALDO (S) | |||
| 26 |
|
0.98 | 0.02 | CB23_PEA (T) | |||
| 27 |
|
0.96 | 0.04 | CB2_PHYPA (T), CB23_POLMU (T) | |||
| 32 |
|
0.65 | 0.33 | 0.02 | CB23_PEA (E) | ||
| 33 |
|
0.02 | 0.98 | CB2_MALDO (V) | |||
| 34 |
|
0.02 | 0.02 | 0.96 | CB2_PHYPA (S), CB2_MALDO (M) | ||
| 42 |
|
0.98 | 0.02 | CB2_MALDO (T) | |||
| 44 |
|
0.02 | 0.98 | CB2_MALDO (K) | |||
| 56 |
|
0.98 | 0.02 | CB2_MALDO (T) | |||
| 57 |
|
0.98 | 0.02 | CB21_WHEAT (Q) | |||
| 58 |
|
0.92 | 0.06 | 0.02 | CB48_MAYZE (R), CB2_PHYPA (T), CB23_ORYSA (T), CB23_PEA (S) | ||
| 59 |
|
0.96 | 0.04 | CB21_PINTH (C), CB21_ORYSA (C) | |||
| 63 |
|
0.98 | 0.02 | CB21_PETSP (E) | |||
| 66 |
|
0.04 | 0.96 | CB25_TOBAC (H), CB21_ORYSA (R) | |||
| 67 |
|
0.98 | 0.02 | CB22_PETSP (T) | |||
| 69 |
|
0.02 | 0.98 | CB22_MAIZE (K) | |||
| 83 |
|
0.02 | 0.98 | CB21_PEA (P) | |||
| 88 |
|
0.98 | 0.02 | CB2_MALDO (R) | |||
| 90 |
|
0.98 | 0.02 | CB2_MALDO (D) | |||
| 91 |
|
0.98 | 0.02 | CB2_MALDO (R) | |||
| 93 |
|
0.98 | 0.02 | CB23_PEA (Q) | |||
| 94 |
|
0.02 | 0.98 | CB21_WHEAT (D) | |||
| 96 |
|
0.84 | 0.14 | 0.02 | CB2_PHYPA (N), CB25_LYCES (S), CB24_LYCES (S), Q38713 (S) |
Tale analisi è più immediata della precedente e permette l'identificazione di ricorrenze particolari come la presenza di Lisina alla posizione 44 (corrispondente al residuo 131, normalmente riportante una Glutammina importante per la coordinazione dei pigmenti, cfr. § B.II.6.1.3 e Figura B-9), ovvero mutazioni con un particolare cambiamento di proprietà aminoacidiche.
In questo caso la sequenza riportante tale mutazione è l'LHC II di melo, una sequenza piuttosto diversa dalla media delle LHC II di tipo I e II, le cui zone di elica sono le seguenti (in grassetto le differenze sostanziali con l'aminoacido corrispondente in pisello sotto-riportato):
elica B elica C
CB2_MALDO PETFAKNRELEVIHSRCAMSAALGCIFPELLSVMG SILAIWTTKVILMGAVEGYR
CB22_PEA W LG RN A Q
elica A elica D
CB2_MALDO TEAFAELKVKELKNGRLAMFSMFGFFVQAIVSRKDRLENLADHLG
CB22_PEA P G GP A
Di particolare rilevanza sono:
Da un'analisi puntuale di questo tipo si è passati ad una più automatica e relativa alle informazioni ricavabili da doppie mutazioni.
Disponendo di un consistente allineamento multiplo e delle informazioni sulle relazioni intercorrenti tra le sequenze (grazie alla visualizzazione come albero filogenetico) è stata studiata la covarianza, o informazione mutuale (cfr. § B.VII), dei residui corrispondenti a quelli di cui si conosce la struttura in LHC II. Il programma utilizzato per le analisi è AliAna (cfr. § C.II.6), creato a questo proposito come parte integrante del lavoro di tesi.Il problema maggiore nell'applicare questo tipo di analisi al caso in esame si è rilevato essere il riconoscimento delle covarianze che possano avere un significato biologico rispetto a quelle determinate dal caso.
La conservazione all'interno della famiglia Lhcb (per le proteine di cui si ha la sequenza) è infatti molto elevata, specialmente considerato il fatto che ci si è concentrati sulle parti dell'allineamento di cui è disponibile la struttura tridimensionale, ovvero le tre eliche transmembrana (B C A) e l'elica terminale anfipatica (D) per un totale di 100 residui.
In queste zone la percentuale di identità aminoacidica è molto elevata (per LHC II tipo I le due sequenze più diverse hanno 77 su 100 aminoacidi identici, con una media di identità a coppie superiore al 90% e 50 residui identici in tutte le sequenze), indicando un'alta importanza di questi aminoacidi nel mantenimento della struttura e della funzione (coordinazione e modulazione dei pigmenti), ma allo stesso tempo abbassando significativamente il numero di coppie covarianti.
A diminuire molto la significatività statistica dell'analisi, contribuisce in particolar modo l'esiguo numero di sequenze disponibili per ogni proteina. Solo trentacinque sequenze per le tipo I, sedici per le tipo II, tre per le tipo III, tre per CP 29, sei per CP 26 e quattro per CP 24.
Il numero di dati ottenibili da questo tipo di analisi - al contempo
- è enorme. Ogni matrice di covarianza è composta da 10000
(1002) elementi, ovvero da tutte le possibili coppie di residui.
Eliminando la diagonale, ovvero le finte coppie formate da un solo residuo
(per cui non vi può essere covarianza) e le coppie ripetute (l'informazione
mutuale è una proprietà simmetrica per cui la covarianza
della coppia di residui i,j è uguale a quella della coppia
j,i), restano comunque 4950 (![]() )
coppie di residui per cui AliAna calcola la covarianza.
)
coppie di residui per cui AliAna calcola la covarianza.
La scelta di un particolare sottoinsieme di sequenze modifica la covarianza (cfr. § B.VII) e quindi ogni nuovo sottoinsieme esaminato produce 4950 misure di informazione mutuale.
Oltre all'insieme totale di 67 sequenze appartenenti alla famiglia Lhcb, sono stati studiati infatti i seguenti sottoinsiemi:
Una stima approssimata del numero di informazioni è quindi di 9 (sottoinsiemi) × 5 (categorizzazioni) × 4950 (coppie per matrice) pari a 222750 misure di covarianza.
Allo stesso modo, anche l'analisi di possibili ponti salini eseguita con lo stesso programma produce una quantità sovrabbondante di dati.
Per rilevare le informazioni significative si è fatto ricorso ad alcuni accorgimenti:
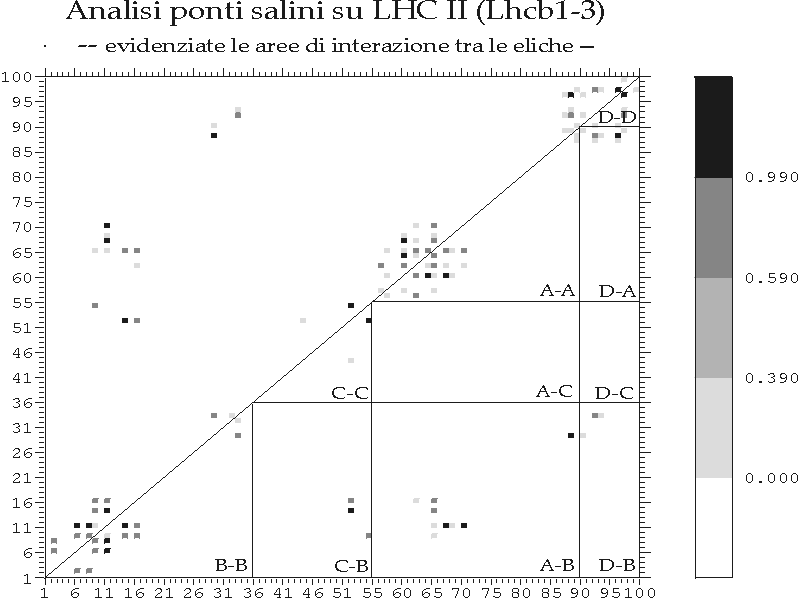 Le matrici appaiono nella seguente
forma grafica:
Le matrici appaiono nella seguente
forma grafica:
Figura -24: Forma grafica delle matrici a punti create in seguito all'analisi del programma AliAna
Ogni quadratino rappresenta una coppia di residui. Il codice colore rappresenta la ricorrenza (nelle sequenze in esame) di coppie di segno opposto che possono formare un ponte salino. È chiara la natura simmetrica delle matrici. Le aree identificate nella precedente figura rappresentano le zone di interazione tra le differenti eliche che compongono la struttura. Interazioni intraelica sono visibili nelle sezioni triangolari mentre quelle interelica sono da ricercarsi nelle zone rettangolari. Per le matrici rappresentanti un'analisi di covarianza, il codice colore rappresenta la misura di covarianza.
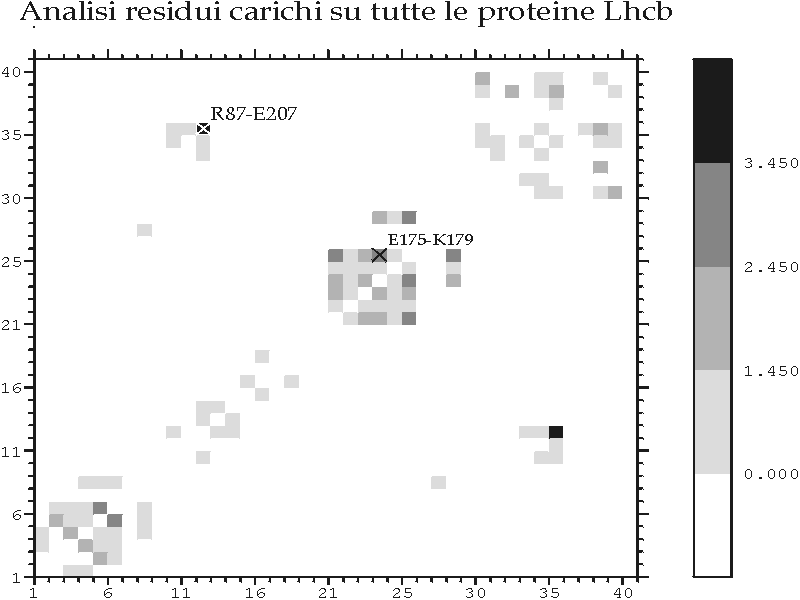 Vengono di seguito riportate
alcune matrici (ridotte per ragioni di chiarezza espositiva e di facilità
di individuazione da 100x100 a 41x41 o 22x22) che evidenziano i risultati
di questa analisi, sotto forma di covarianza per alcune coppie di residui
che possono dare ponti salini (cfr. § B.VIII.1), sulla cui individuazione
ci si è concentrati.
Vengono di seguito riportate
alcune matrici (ridotte per ragioni di chiarezza espositiva e di facilità
di individuazione da 100x100 a 41x41 o 22x22) che evidenziano i risultati
di questa analisi, sotto forma di covarianza per alcune coppie di residui
che possono dare ponti salini (cfr. § B.VIII.1), sulla cui individuazione
ci si è concentrati.
Figura -25: Analisi di covarianza per residui ionizzabili sulla famiglia Lhcb
(vedi didascalia Figura D-24)
La figura precedente è il risultato di un'analisi mirata ai residui carichi (usando la categorizzazione aminoacidica "CHRG" di AliAna, cfr. § C.II.6) sull'insieme totale di proteine Lhcb. Due coppie di residui con alta covarianza vengono indicate, corredate dalla corrispondente coppia aminoacidica ritrovata nella proteina LHC II cristallizzata.
Figura -26: Analisi di covarianza su un sottoinsieme rappresentativo delle proteine Lhcb
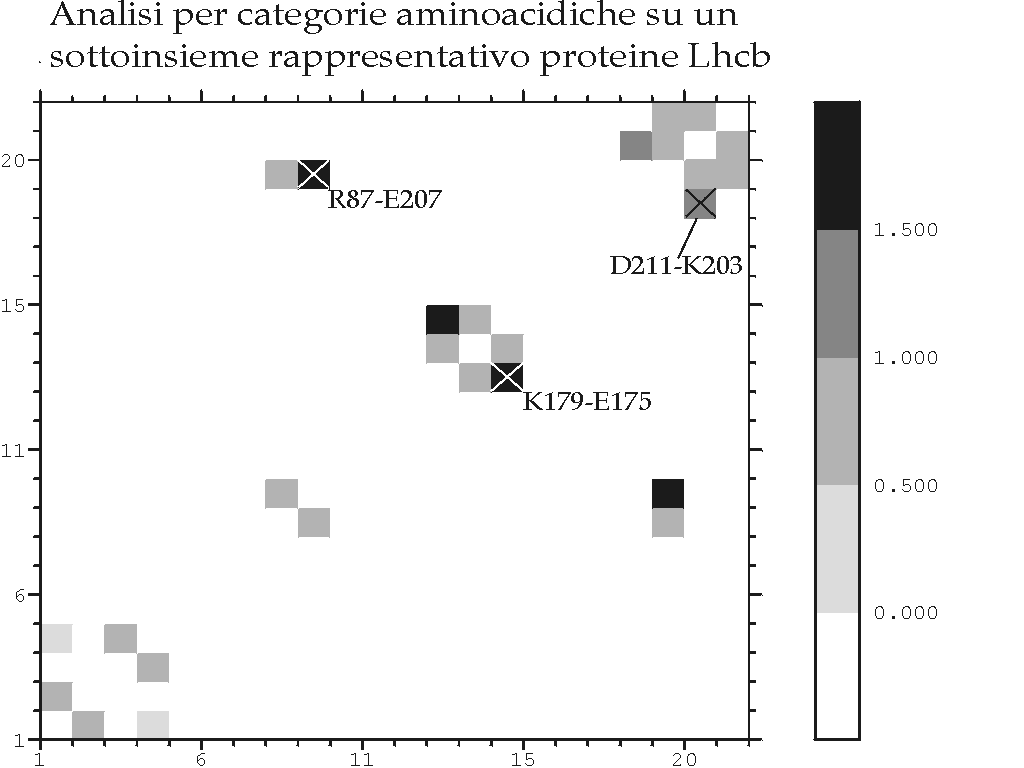
In questo caso l'analisi è condotta per categorie aminoacidiche
(categorizzazione "FIV") sul sottoinsieme rappresentativo. Sono indicate
tre coppie importanti (una in più rispetto all'analisi precedente).
Le tre coppie sono ora più distinguibili dalle altre, essendo le
uniche con covarianza maggiore di 1. L'aver rimosso una gran parte di sequenze
molto omologhe tra di loro (in particolare la sovrabbondanza delle LHC
II di tipo I) ha di fatto aumentato la sensibilità dell'analisi
di covarianza. Le tre coppie appartengono tutte a ponti salini probabili
in LHC II ma che sono assenti, conservate o invertite nelle antenne minori,
come mostrato dalla seguente tabella:
|
(in grassetto i possibili ponti salini conservati, in corsivo se assenti) |
|||||
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
da cui si vede che le risposte fornite da questo tipo di analisi non sempre sono chiaramente interpretabili.
Figura -27: Analisi di covarianza per residui ionizzabili sulle proteine LHC II di tipo I e II
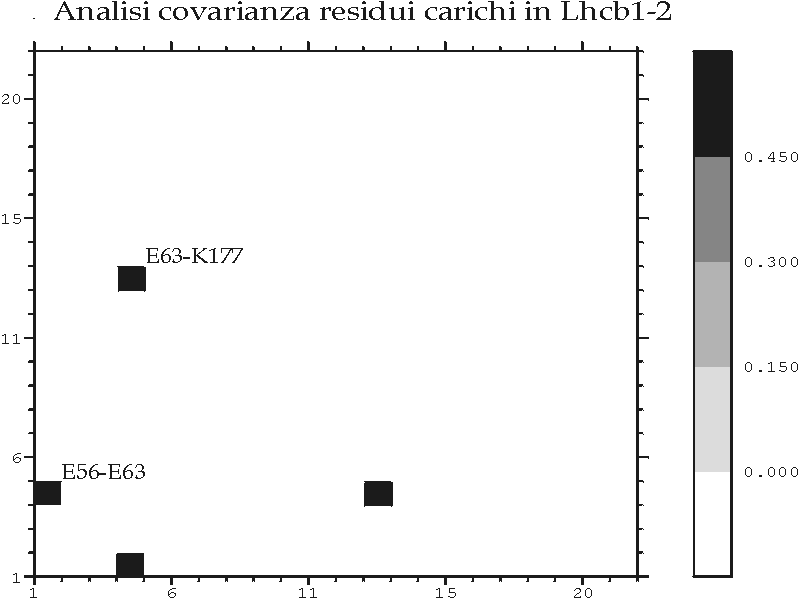
Questa figura evidenzia l'estrema carenza di informazione mutuale. L'analisi è condotta per un sottoinsieme comprendente solo le LHC II di tipo I e II, per un totale di 51 sequenze, al fine di studiare le covarianze all'interno della stessa proteina (nei diversi organismi) e non quelle derivanti dalla differenziazione proteica con le antenne minori. La conservazione di queste sequenze è tale da portare alla luce solo due coppie. In questo caso si assiste ad un'inversione di segno ovvero alla presenza di sequenze che abbiano K63-E177 invece che E63-K177. Questo aumenta grandemente la plausibilità di un'interazione ponte salino tra i due residui presenti alle posizioni 63 e 177 dato che la doppia mutazione ristabilirebbe il legame.
La presenza nella matrice della coppia di residui 56-63 è collegata alla coppia 63-177. Infatti nei casi in cui E63 è sostituito da K63, la posizione 56 muta da E ad A (cfr. anche § D.VI).
Purtroppo, come detto sopra, l'esiguità del campione (il basso numero di sequenze) e l'alta conservazione aminoacidica tra le sequenze (nonché le difficoltà nell'individuazione dei dati significativi) portano alla luce poche coppie di residui covarianti dotate di un possibile ed intuitivo significato biologico.
Comunque queste informazioni, unite ai risultati della dinamica molecolare ed al confronto delle sequenze, permettono di ampliare il punto di vista ed estendere le conoscenze sulle possibili interazioni chiave per la struttura proteica.
Le possibili coppie aminoacidiche formanti ponte salino, individuate dall'analisi di covarianza e dalla dinamica molecolare, di cui si parlerà più avanti (cfr. § D.VIII) nella formulazione di ipotesi di modello, sono riportate nella seguente figura, prodotto dell'analisi di possibili ponti salini effettuata con AliAna:
Figura -28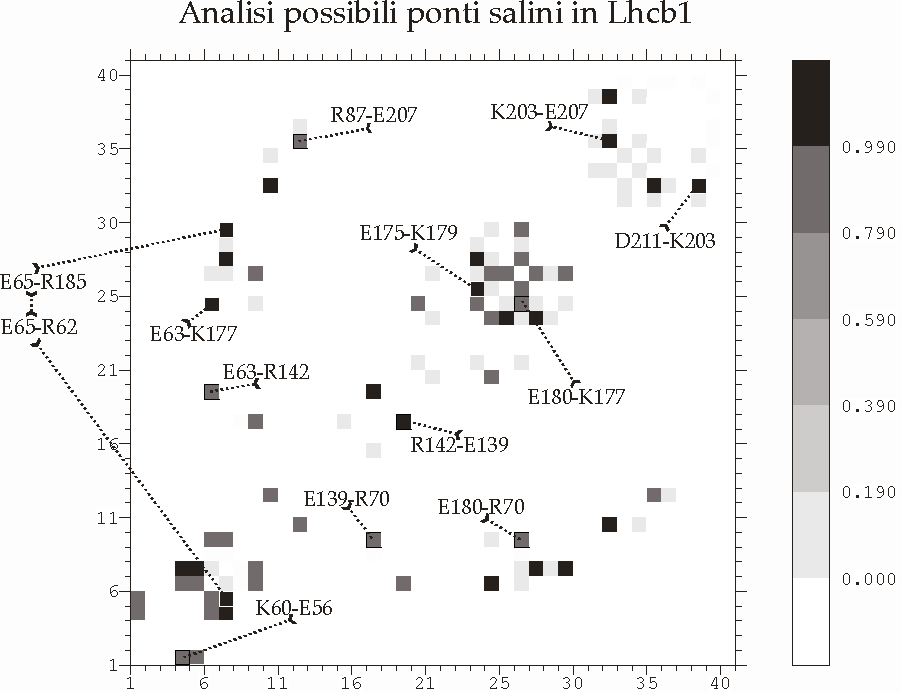 :
Analisi dei possibili ponti salini in LHC II tipo I
:
Analisi dei possibili ponti salini in LHC II tipo I
Il protocollo di simulated annealing approntato (cfr. § D.IV.1.2) è stato ad essa applicato per studiare eventuali interazioni tra le catene laterali delle eliche transmembrana delle diverse subunità, alla ricerca di possibili contatti importanti per la conformazione trimerica.
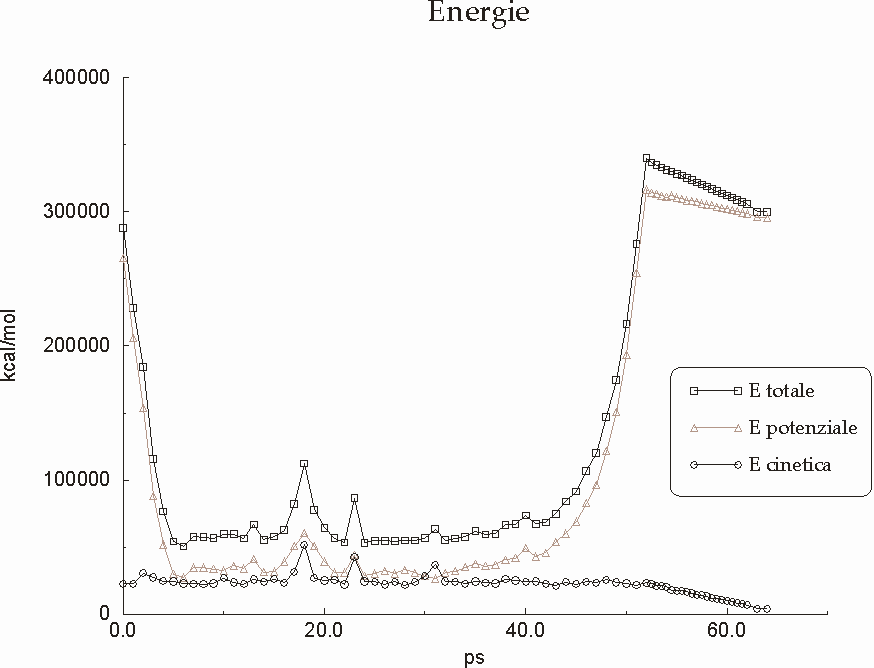 Seguono i grafici relativi
alla dinamica molecolare:
Seguono i grafici relativi
alla dinamica molecolare:
Figura -29: Andamento delle energie nel corso della simulazione del trimero
Figura -30: Andamento delle temperature nel corso della simulazione del trimero
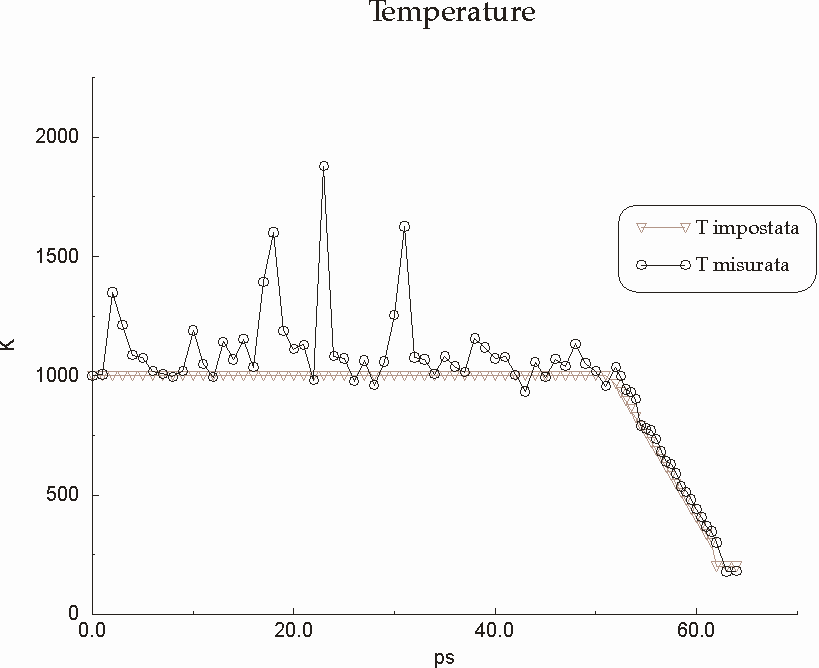 (temperatura
impostata nel protocollo e temperatura misurata durante la simulazione)
(temperatura
impostata nel protocollo e temperatura misurata durante la simulazione)
È possibile notare dal grafico delle energie (Figura D-29) che i valori sono minori od uguali al triplo dei valori rilevati nella simulazione con il monomero e non maggiori.
Tale comportamento indica che non vi sono interazioni sfavorevoli (che porterebbero l'energia a livelli maggiori del triplo) e che invece la presenza di interazioni attrattive di van der Waals tra le subunità diminuisce l'energia totale del sistema.
La struttura del trimero indica come possibili aree di interazione tra parti polipeptidiche dei diversi monomeri (limitatamente alle zone d'elica presenti nella struttura cristallografica):
Figura -31: Rappresentazioni della struttura ricostruita del trimero
Visione perpendicolare - dal lato stromatico - (in alto) e parallela al piano della membrana (in basso)
Si riconosce un importante ruolo per la trimerizzazione di LHC II alla porzione C-terminale della proteina.
Uno studio di delezioni progressive mostra come la rimozione di aminoacidi fino a 10 dal C-terminale non modifica significativamente la stabilità del complesso mentre quando l'undicesimo (Triptofano 222) è rimosso viene compromessa la stabilità dei singoli monomeri [Paulsen e Kuttkat 1993].
La sostituzione del singolo Trp 222 con Istidina, Alanina o Glicina abolisce completamente l'abilità della proteina di riassemblarsi trimericamente che invece non è influenzata dalla sostituzione con Fenilalanina [Kuttkat et al. 1996].
Inoltre un motivo legato alla trimerizzazione è stato identificato nella sequenza 'WYGPDRVKYL' presente all'N-terminale della proteina (aminoacidi 16-25 in LHC II di pisello). La sua (parziale) rimozione o la sostituzione di alcuni dei suoi aminoacidi impedisce la ricostituzione in trimeri di LHC II [Hobe et al. 1995].
Lo studio comparato delle regioni N-terminali nelle sequenze dei geni Lhcb porta alle seguenti osservazioni su alcuni fattori che potrebbero essere coinvolti nella trimerizzazione di LHC II, mancanti nelle antenne minori:
S,A L L
WYGPDRVKYLGPFSG LHC II tipo II
E,A PLF
WYGPDRVKYLGPFS- LHC II tipo III
WFPGAV--------- CP 29
Y I
WYGPDRRIYLPDGLL CP 26
F N,E
WIPAVKGGGNLV--- CP 24
IRSDAEI
pphphhph-hhh-hhh-phhhph-h-hhh+p+ LHC II tipo III
ph-hh-hphhh-hhh-hhhhh+hhhhhp+h+ CP 29
-hh-hhph-hhh-hhh-hhhhh++h-phh+hp CP 26
h-h-hh-hphhh-hhh-hhhhh+-hhhh+hh+
CP 24
Questo potrebbe essere indice dell'importanza di interazioni polari nella formazione e stabilità del complesso trimerico LHC II, con possibili ponti idrogeno tra le catene laterali dei loop N-terminali dei tre monomeri.
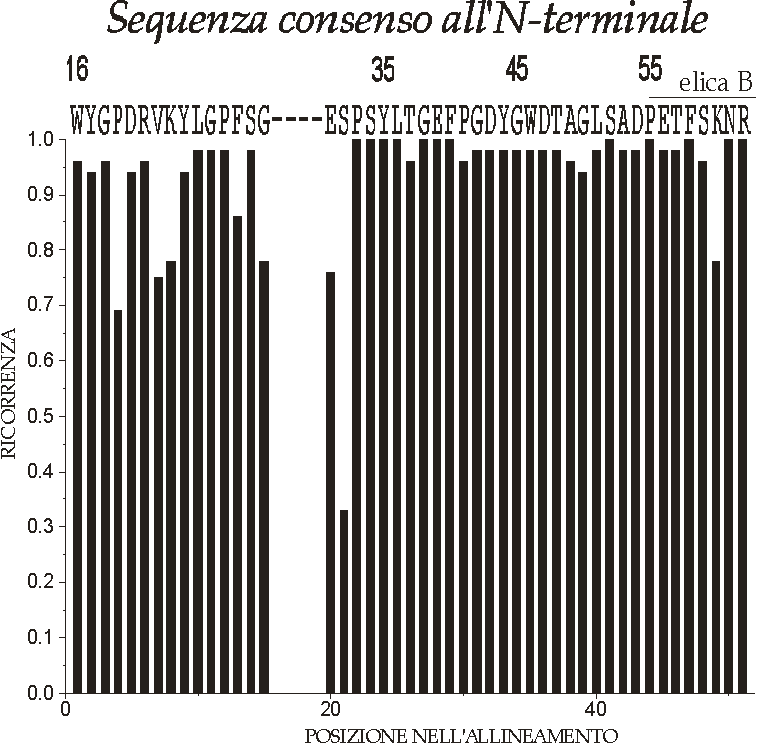 La sequenza consenso per la
porzione N-terminale, con le ricorrenze osservate, per le proteine LHC
II di tipo I e II:
La sequenza consenso per la
porzione N-terminale, con le ricorrenze osservate, per le proteine LHC
II di tipo I e II:
Figura -32: Ricorrenza aminoacidica e sequenza consenso N-terminale per LHC II tipo I e II
Segue un'analisi simile condotta per la porzione C-terminale a partire dall'aminoacido His212 fino al termine delle sequenze che evidenzia la divergenza di questa zona tra le antenne minori e i polipeptidi del complesso LHC II (h=idrofobici, p=polari, -=acidi, +=basici; in grassetto sottolineato le differenze con Lhcb1-2):
+hh-hhppphhhhhpphhhh+ LHC II tipo I e II
+h--hhhpphhhhhp+hhhhh LHC II tipo III
+hp-hh+pphh-phhhpp CP 29
+hp-hhhpphhphhphhh-+phph CP 26
assente elica D e loop C-terminale CP 24
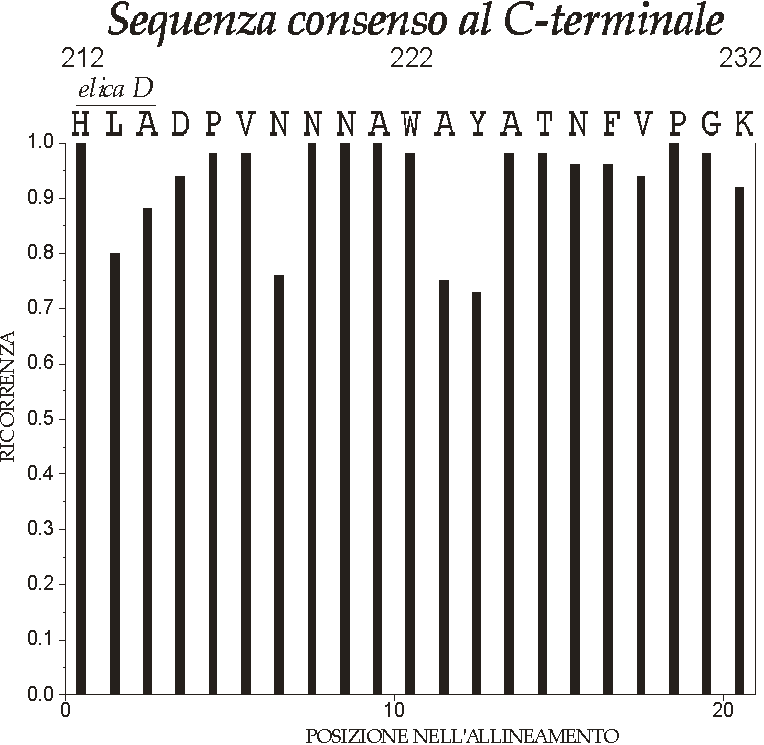 La sequenza consenso per LHC
II tipo I e II è indicata nella seguente figura:
La sequenza consenso per LHC
II tipo I e II è indicata nella seguente figura:
Figura -33: Ricorrenza aminoacidica e sequenza consenso C-terminale per LHC II tipo I e II
La struttura risultante dalla simulazione infatti mostra un doppio ponte tra l'elica C e l'elica B invece della singola coppia ionica intraelica al termine dell'elica C.
Questo indicherebbe che tale disposizione spaziale degli aminoacidi è possibile e addirittura preferibile in termini energetici alla coppia interna - almeno nella situazione di simulazione che, lo ricordiamo, è mancante di vari oggetti potenzialmente fondamentali per la sua correttezza.
Da qui si è partiti con una serie di studi da più direzioni per elaborare un modello per la proteina, che potesse anche non coinvolgere necessariamente le coppie ioniche ipotizzate da Kühlbrandt et al. [1994], e stabilirne la plausibilità.
Oltre alla simulazione di dinamica molecolare (che rappresenta lo sfruttamento del dato spaziale) si è ricorsi allo studio accurato delle sequenze aminoacidiche della famiglia multigenica comprendente LHC II e le antenne minori (CP 29, CP 26, CP 24), sfruttando quindi il dato di sequenza.
Per questo fine è stato elaborato un programma polivalente di analisi di allineamento AliAna che potesse estrarre da un allineamento multiplo informazioni quali la covarianza aminoacidica (doppi mutanti), la possibilità di ponti disolfuro, la possibilità di ponti salini semplici (cfr. § C.II.6).
Lo studio della covarianza identifica possibili coppie cariche - anche molto distanti nella sequenza aminoacidica - interagenti in ponte salino e dà ad esse una certa plausibilità.
L'analisi delle sequenze porta alla luce esempi di sequenze mutanti per un aminoacido chiave (in relazione ad un modello ipotizzato) mentre praticamente identiche per tutti gli altri residui. In particolare alcune sequenze riportano la mancanza del residuo E180, che nel modello ipotizzato da Kühlbrandt et al. è di particolare importanza per il mantenimento della struttura, essendo coinvolto in un ponte salino interelica (cfr. il modello 1 nella trattazione seguente).
Sono inoltre disponibili alcuni esperimenti di mutagenesi sito-specifica effettuati su LHC II e su CP 29 (una delle antenne minori) i cui risultati, soprattutto per quanto riguarda la resa di ricostituzione in relazione alla posizione aminoacidica mutata, offrono altri tasselli per la comprensione del mosaico.
Ci si è concentrati soprattutto sull'elica C e su possibili interazioni di questa con le altre due eliche. La struttura identifica chiaramente la posizione dell'elica C, anche alla bassa risoluzione di 3.4 Å.
Questo implica che la sua posizione sia ben delineata, fissa ed invariante nel cristallo a differenza dei "loop" o delle catene fitoliche che non appaiono nella struttura tridimensionale. Ma la struttura mostra anche che l'elica C è alquanto distante dalle altre due eliche (la distanza minima fra atomi dello scheletro polipeptidico è di 10 Å dall'elica B e 18 Å dall'elica A), che formano invece una struttura unita e stabile.
Tra l'elica C e le altre due eliche si forma una tasca idrofobica accomodante 5 clorofille (siti B5, B6, B1, A6 e A7) [Kühlbrandt et al. 1994]. Vi è sicuramente una forte interazione tra le catene fitoliche di queste che così forniscono un saldo legame, essendo anche al centro della membrana, nell'ambiente più idrofobico.
Studi di ricostituzione proteica mostrano che in effetti sarebbe proprio la sinergia tra catene polipeptidiche e pigmenti l'origine della corretta ricostituzione della proteina. In LHC II il refolding avviene solo in presenza di tutti i pigmenti - almeno in vitro [Booth e Paulsen 1996] (cfr. anche § B.II.6.1.3 per il ruolo strutturale attribuito alle xantofille).
Studi in CP 26 indicano invece come siano indispensabili solo le due luteine o, in misura minore, altri carotenoidi [Ros et al. 1998].
La ricerca di eventuali ulteriori ragioni per cui l'elica C si trovi in tale posizione ha portato alle seguenti riflessioni ed ipotesi.
I loop colleganti le eliche transmembrana potrebbero avere una struttura non organizzata o poco strutturata, non risultando nella struttura cristallografica. Non apparirebbero essere questi dunque i responsabili della locazione e orientazione di tale elica.
L'unica interazione di tipo idrofobico tra le eliche A e B con l'elica C si può avere tra gli aminoacidi Trp71 (in B) e Val138 o Met135 (entrambi in C).
Questa non potrebbe da sola impedire la fluttuazione o vibrazione dell'elica C che comprometterebbe la sua "visibilità" alla diffrazione degli elettroni.
L'attenzione si sposta quindi sulle coppie ioniche (molto rilevanti perché questa è una proteina transmembrana e perché gli esperimenti di mutagenesi mostrano come esse siano fondamentali per la ricostituzione della proteina) e permette l'elaborazione di tre diversi modelli di cui di seguito si daranno i pro ed i contro.
E180-R70
E63-K177
E63-R142
E180-K177
E180-R70
E56-K60
E175-K179
D211-K203
E207-R87-E83 (formanti un ponte salino complesso)
oppure
E207-R87 (formanti ponte salino semplice ed un ponte idrogeno tra Ossigeno di E83 ed Azoto dello scheletro polipeptidico di E207)
E56-K60 e E175-K179 potrebbero essere ponti salini interni che stabilizzano l'elica compensandone il dipolo (cfr. § B.VIII.3) per la presenza dell'aminoacido acido in posizione N-terminale.
E207-R87 sono posizioni ugualmente occupate da aminoacidi neutri nelle proteine di alghe mentre la coppia D211-K203 ha una corrispondente coppia con inversione di segno in CP 26 (K-E), aumentando fortemente la plausibilità di un ponte salino tra questi due aminoacidi. Queste due insieme possono spiegare l'orientazione dell'elica anfipatica D, la quale accomoda la testa di una luteina.
Nella pagina seguente:
Figura -34: Rappresentazione della struttura dei due ponti salini complessi ipotizzati
In alto: R62-E65-R185 In basso: E207-R87-E83 e, sulla sinistra, D211-K203
Queste due proteine sicuramente non possono avere il ponte salino interelica E180-R70 e lascerebbero quindi R70 disaccoppiata.
Termodinamicamente questo è molto sfavorevole e quindi improbabile in un ambiente idrofobico come il centro della membrana.
A favore di tale modello vi sono però:
se pure il risultato della mutazione E139/R142 (comportante la perdita di 5 clorofille e 1-2 carotenoidi ed un drastico calo al 40% della resa di ricostituzione) è spiegabile come perdita di stabilità dell'elica C derivante dalla scomparsa del ponte intraelica, lo stesso effetto si dovrebbe riscontrare con le singole mutazioni se l'effetto fosse determinato solo dalla perdita di tale ponte.
Invece mutando solo E139 con una Leucina si ha una resa di ricostituzione dell'83% e la perdita di 1.5 clorofille.
In CP29, inoltre, la mutazione singola corrispondente a R142 impedisce la ricostituzione della proteina, allo stesso modo della doppia mutazione degli aminoacidi corrispondenti a E139 e R142. La mutazione corrispondente a E139 comporta invece poco meno di metà proteine non ricostituite.
Confrontando la posizione dei due residui coinvolti è possibile spiegare questa differenza nell'influenza delle due mutazioni singole:
Nella pagina seguente:
Il modello 2, ipotizzato come il più probabile dalla simulazione
di dinamica molecolare, si scontra con alcuni dati derivanti dagli esperimenti
di mutagenesi sito-specifica: la mutazione del residuo che in CP 29 equivale
all'R70 di LHC II, con un residuo idrofobico, non dà ricostituzione
proteica, mentre la mutazione del residuo corrispondente a E139 (con un
aminoacido idrofobico) influenza solo per metà circa la ricostituzione.
In LHC II la mutazione della singola R70 non è stata eseguita mentre
E139L dà 83% di ricostituzione. Se effettivamente questi due aminoacidi
fossero correlati e se la perdita del ponte salino fosse causa di una destabilizzazione
della struttura proteica come ipotizzato, ci si aspetterebbe un'eguale
resa di ricostituzione per le due mutazioni (è comunque possibile
che in CP 29 l'organizzazione dei ponti salini segua il modello 1 mentre
in LHC II segua il presente modello 2, cfr. § D.IX.1).
Questo modello spiegherebbe la posizione relativa dell'elica C grazie ad un forte legame tra essa e l'elica B derivante dai due ponti salini ipotizzati.
La doppia mutazione "E139L/R142L" che dà solo il 40% di resa ricostituzione in LHC II provocherebbe la rottura dei due ponti salini, con la comparsa di cariche spaiate termodinamicamente molto sfavorite. Tale mutazione comporta la perdita di ben 5 clorofille e di 1-2 carotenoidi. Ovvero una distruzione praticamente completa della tasca idrofobica che alloggia questi pigmenti.
Il modello 1 spiega questo con la perdita del ponte salino intraelica comportante una possibile destabilizzazione dell'elica; il presente modello reputa invece responsabile l'allontanamento (o comunque una riorientazione) dell'elica C.
Nella pagina seguente:
Figura -36: Rappresentazione del modello 2 (in alto senza ed in basso con le clorofille)
Il modello 3 darebbe una spiegazione alternativa a quella sopra
esposta (cfr. § D.VIII.1) per la differenza intercorrente tra le singole
mutazioni di Glu139 e Arg142 nell'influenzare drasticamente la resa di
ricostituzione ed il numero di pigmenti coordinati dalla proteina, come
evidenziato dalla seguente tabella:
| Proteina | Sito/i mutato/i | Resa di ricostituzione | Clorofille perse |
| LHC II | E139 |
|
|
| CP29 | E174 (E139 in LHC II) |
|
|
| LHC II | R142 |
|
|
| CP29 | R177 (R142 in LHC II) |
|
|
| LHC II | E139 R142 |
|
|
| CP29 | E174 R177 (E139 R142) |
|
Però non vi sono dati per rispondere alla domanda "cosa compensa la carica del Glutammico 139 permettendogli di coordinare clorofilla?".
È stato dimostrato che Glu può essere un ligando per clorofilla ma questo in congiunzione con la presenza di un catione Ca++ che farebbe coppia con l'aminoacido e permetterebbe la coordinazione. Questo è stato dimostrato in CP29 per quanto riguarda il residuo E166 (corrispondente in LHC II a Q131) [Jegerschöld et al., in preparazione].
Inoltre la mutazione Q131E (Gln® Glu) in LHC II non comporta perdita di clorofilla [Varotto, tesi di laurea].
Se il modello 3 fosse quello più vicino alla realtà dovrebbe
esserci un catione positivo a compensazione della carica del residuo Glutammico.
Non vi sono dati a riguardo.
Nella pagina seguente:
Figura -37: Rappresentazione del modello 3 (in alto senza ed in basso con le clorofille)
-----
Un altro dato interessante, comune ai tre modelli, derivante dall'analisi delle sequenze è il seguente:
LHC II di cetriolo (CB22_CUCSA), proteina con 97% identità e 98% analogia per quanto riguarda le eliche transmembrana con LHC II di pisello, ha una Treonina in posizione 70 invece dell'apparentemente molto importante Arginina, senza altre modificazioni a questo connesse (cfr. § D.VI).
Potrebbe essere la formazione di un ponte idrogeno tra questa Treonina e l'eventuale residuo Glutammico coinvolto in ponte salino a sostituire strutturalmente l'ipotizzato legame di natura elettrostatica, ma la carica resterebbe non compensata.
Non si può escludere che le proteine appartenenti alla famiglia multigenica adottino diverse configurazioni di ponti salini (per alcune proteine alcuni modelli non sono possibili data la mancanza di residui chiave) e che questo possa essere causa di differenze nella loro struttura che pur non essendo sostanziali conferiscano alle proteine diverse funzioni (ad esempio un differente numero di clorofille legate o diverse orientazioni delle stesse).
A nostro avviso le proteine LHC II e CP 24 adotterebbero la configurazione proposta nel modello 2 mentre CP 29 e CP 26 adotterebbero quella proposta nel modello 1, come delineato ulteriormente nel prossimo capitolo (§ D.IX).
Allo scopo di discriminare tra i differenti modelli qui formulati, si suggerisce la possibilità di pianificare esperimenti di mutagenesi sito-specifica a questo mirati, tramite doppie mutazioni con inversioni di segno degli aminoacidi carichi costituenti un ponte salino nel modello che si intende provare od escludere.
Ad esempio le mutazioni:
E180R | R70E
R70E | E139R
In questo modo se effettivamente vi fosse un ponte salino tra i due aminoacidi, questo non verrebbe ad essere sostanzialmente modificato (anche se queste mutazioni potrebbero influenzare in senso negativo la stabilità proteica). Se invece i due aminoacidi fossero coinvolti in due distinti ponti salini questo comporterebbe l'assenza di entrambi i ponti salini nella proteina ricostruita in vitro con i pigmenti.
Le tre antenne minori (CP29, CP26 e CP24) sono state modellate per
omologia (cfr. § B.VI) alla struttura di LHC II.
Alcune differenze significative vengono qui delineate, soprattutto in relazione alle ipotesi di modello di cui si è discusso nel capitolo precedente (la numerazione usata è sempre quella relativa alla LHC II di pisello e si rimanda alla tabella § F.II in appendice per la corrispondenza con la numerazione delle antenne minori).
Per la proteina CP 29 il modello 2 appare poco probabile poiché
la sostituzione di una Glutammina alla Lisina 177 lascia la carica del
Glutammico 180 non compensata.
Per lo stesso motivo il modello 1 potrebbe essere modificato dalla coppia interelica E63-K177 alla coppia intraelica E63-R60 che però non apporta un contributo stabilizzante in relazione al dipolo dell'elica.
Le coppie comuni ai tre modelli ipotizzate per LHC II sono in CP29 assenti ma dalla struttura si può rilevare la possibilità di un ponte idrogeno tra una Lisina in posizione 203 ed una Treonina in posizione 211 che potrebbe orientare la corta elica D.
I dati biochimici indicano che questa proteina coordina solo 8 clorofille e gli esperimenti di mutagenesi [Simonetto, tesi di laurea] hanno identificato i siti coordinati: a1, a2, a3, a4, a5, b3, b5 e b6 (vedi schema pigmenti, Figura B-9).
Le clorofille mancanti sarebbero b2, b1, a6 e a7, che - con l'eccezione di b2 - appartengono alla tasca idrofobica tra l'elica C e le altre due eliche.
Nota: CP 29 coordina sei clorofille di tipo a e due di tipo b. Si è scelto per semplicità di trattazione di mantenere la nomenclatura di Kühlbrandt et al. per indicare le clorofille (cfr. § B.II.6.1.3) anche se, applicata a CP 29, essa appare in contrasto con i dati biochimici (lasciando ad intendere che CP 29 coordini cinque clorofille di tipo a e tre di tipo b).
Si ipotizza qui che un'orientazione diversa (rispetto a LHC II) dell'elica C - forse data dal diverso modello di configurazione dei ponti salini adottato - potrebbe essere alla base di queste differenze nella coordinazione dei pigmenti.
Altre differenze risiedono:
Come per CP 29 potrebbe essere una diversa disposizione dell'elica C in relazione alle altre due la ragione della minore coordinazione in pigmenti dato che i ligandi ipotizzati da Kühlbrandt et al. [1994] per 9 clorofille sono conservati in CP26 (ad eccezione della mutazione Gln® Glu in posizione 131 che potrebbe comunque coordinare clorofilla come visto in CP 29 [Simonetto, tesi di laurea] e LHC II [Varotto, tesi di laurea]) con la possibile interazione con uno ione positivo.
Le clorofille a7 e b1 vengono infatti perse in LHC II in seguito a mutazione degli aminoacidi dell'elica C (mutazione E139L/R142L: perdita di clorofille b5, b6, b1, a6, a7; mutazione Q131L: perdita di clorofille 'b6' e 'a6 o a7' [Remelli, tesi di laurea]).
Supponendo che le clorofille coordinate da CP 29 e CP 26 siano le stesse, una spiegazione alla coordinazione di una clorofilla in più da parte di CP 26 potrebbe essere la seguente:
CP 29 ha una Valina in posizione 82. Questo fatto implicherebbe la presenza del ponte idrogeno tra i residui nelle posizioni 78 e 82 e quindi una non disponibilità del carbonile peptidico al residuo 78 per la coordinazione della clorofilla.
CP 29 verrebbe quindi ad avere una clorofilla in meno rispetto a CP 26 per la presenza della Valina 82 e viceversa CP 26 una in più per la presenza di Prolina in tale posizione.
In relazione ai ponti salini ipotizzati comuni ai tre modelli, CP 24 non può avere - al pari di CP 29 - le due coppie E56-K60 e E175-K179 a causa della sostituzione degli aminoacidi in queste posizioni, né vi possono essere le coppie relative all'elica D, per l'assenza di questa.
I dati biochimici indicano la coordinazione di 10 clorofille per CP 24, aumentando le somiglianze strutturali con LHC II nonostante la minor omologia di sequenza.
Due rilevanti differenze nei ligandi per le clorofille ipotizzati da Kühlbrandt et al. [1994] sono:
Le due clorofille in meno che CP 24 coordina rispetto a LHC II potrebbero quindi essere b3 (per l'assenza del residuo His 212) e a6 per le ragioni esposte nel paragrafo precedente (§ D.IX.2) in relazione alla coordinazione con un carbonile dello scheletro polipeptidico (CP 24 ha una Glicina in posizione 82 invece che una Prolina come CP 26 e LHC II).
Nella pagina seguente:
L'approccio bioinformatico con cui si è deciso di lavorare
evidenzia in questo lavoro di tesi l'importanza dell'interconnessione tra
la sperimentazione e la modellistica.
Le analisi che in questa tesi confluiscono nei modelli per la proteina LHC II, e per le antenne minori a lei omologhe, rivelano la possibilità che una diversa organizzazione di alcuni aminoacidi chiave nel mantenimento della struttura sia alla base delle differenze funzionali tra tali proteine.
CP 26 e CP 29 hanno infatti caratteristiche simili che le differenziano
da LHC II e CP 24 come riassunto nel seguente schema:
|
|
|
|
|
|
| Clorofille coordinate |
|
|
|
|
| Xantofille coordinate |
|
|
|
|
| Presenza di zeaxantina legata2 |
|
|
|
|
| Presenza Glu in posizione 1314 |
|
|
|
|
1: per monomero
2: xantofilla implicata nel processo di quenching dell'energia luminosa in eccesso
3: dato non certo
4: la protonazione di questo residuo causerebbe una riorganizzazione dei pigmenti nella porzione proteica prossimale al lumen, responsabile del quenching non fotochimico
In particolare la presenza di zeaxantina e di un residuo Glutammico in posizione 131 ed alcuni esperimenti condotti in questa direzione identificherebbero le proteine CP 29 e CP 26 come implicate nel processo di regolazione del trasferimento di energia luminosa tramite dissipazione dell'eccesso di energia sotto forma non radiativa (calore) [Bassi et al. 1997; Sandonà et al. 1998].
La posizione occupata da questi complessi nell'arrangiamento proposto per il fotosistema II (vedi Figura B-8) e la stechiometria di questi in relazione al centro di reazione (una proteina CP 26 e una CP 29 per ogni centro di reazione) rappresentano un'ulteriore differenza con CP 24 (in posizione periferica con stechiometria non accertata) ed LHC II (in quantità molto maggiore e con la possibilità di migrare verso il fotosistema I, cfr. § B.II.6.1).
Lo studio di modellistica realizzato in questo lavoro di tesi propone l'ipotesi che alla base di tali differenze funzionali (o di alcune di esse) ci possa essere una diversa organizzazione di alcuni ponti salini importanti per il mantenimento della struttura proteica. In particolare si ipotizza un modello per LHC II e CP 24 con un doppio legame di natura elettrostatica tra l'elica C e l'elica B, assente nel modello relativo a CP 29 e CP 26.
I ponti salini proposti da Kühlbrandt et al. [1994] - descritti nel modello 1 - sarebbero quindi quelli presenti nelle due antenne minori CP 29 e CP 26.
Gli esperimenti di mutagenesi sito-specifica condotti in CP 29 sono infatti in perfetto accordo con tale modello mentre gli esperimenti su LHC II, in cui vengono mutati gli aminoacidi ionizzabili appartenenti all'elica C, sarebbero meglio spiegati dal modello 2 qui proposto, con i due ponti salini tra le eliche C e B.
L'approccio bioinformatico utilizzato non può comprovare o negare le ipotesi formulate ma fornisce la possibilità di una razionalizzazione nella pianificazione di esperimenti che possano convalidare o confutare i modelli qui esposti.
-----
Uno studio su tutte le sequenze disponibili dei prodotti dei geni Lhcb, correlate con un alto grado di omologia alla proteina LHC II, e le simulazioni molecolari sulla struttura ricostruita confluiscono nei modelli presentati per la proteina LHC II e per le antenne minori CP 29, CP 26 e CP 24.
L'allineamento delle proteine antenna codificate dai geni Lhcb è stato esteso a comprendere tutte le sequenze di questo tipo attualmente disponibili. Questo ha permesso di studiare la conservazione di singoli aminoacidi all'interno della famiglia multigenica e di compiere studi sulla variazione contestuale di coppie di residui, per mezzo di un programma scritto a tale proposito.
Alcune informazioni di possibile interesse strutturale sono state quindi estratte a partire dalle sequenze, soprattutto in relazione ai ponti salini che appaiono molto importanti per il mantenimento della corretta struttura terziaria e per la coordinazione dei pigmenti.
Si è cercato di ricostruire alcune parti mancanti della struttura cristallografica di LHC II partendo dalla posizione dei soli atomi Ca delle eliche transmembrana e dalla posizione dei pigmenti che questa proteina coordina. Un algoritmo automatizzato posiziona le catene laterali e ricostruisce lo scheletro polipeptidico a partire dai Ca ma non tiene in considerazione la presenza dei pigmenti.
Per fornire una disposizione teoricamente più esatta delle catene laterali, è stata applicata alla struttura la metodica di simulated annealing con rilassamento delle costanti di forza di Nilges et al. [1988]. Questo ha permesso di superare le limitazioni del normale simulated annealing e di ottenere una conformazione delle catene laterali che non fosse influenzata dalla disposizione automatizzata.
La simulazione molecolare ha mostrato come possibile una configurazione di ponti salini diversa da quella proposta da Kühlbrandt et al. [1994] per LHC II.
La combinazione dei risultati forniti dalla simulazione molecolare e dallo studio delle sequenze è stata confrontata con i dati biochimici ed ha permesso la formulazione dei modelli presentati per le quattro proteine omologhe.
-----
Il campo di forze CVFF è stato esteso con i parametri che permettono la simulazione di tetrapirroli di clorofilla e rende possibili ulteriori studi di simulazione sulle proteine leganti clorofilla ed il raffinamento, sfruttando tale campo di forze, di nuove strutture che contengano questo pigmento.
Il programma AliAna realizzato permette vari tipi di analisi per allineamenti multipli di una famiglia di sequenze omologhe e l'integrazione di informazioni strutturali - anche parziali - all'analisi delle sequenze: la conoscenza della posizione dei soli Ca può essere utilizzata per esaminare possibili interazioni molecolari al livello delle sequenze.
È stato realizzato un protocollo di indagine bioinformatica, integrante studio di sequenza e simulazione molecolare, che può essere applicato ad altri sistemi, dovunque vi sia una proteina con struttura non completamente risolta ed un numero sufficiente di sequenze ad essa omologhe. Esso offre la possibilità di una ricostruzione plausibile delle catene laterali e l'integrazione dello studio di sequenza al normale modelling per omologia.
In conclusione, sono stati applicati diversi metodi di analisi di sequenza e di simulazioni molecolari al fine di analizzare, in maniera il più possibile estensiva, le implicazioni strutturali derivanti dall'arrangiamento dei Ca ottenuti mediante cristallografia elettronica e dalla naturale occorrenza di certi residui, o coppie di residui, in alcune posizioni di sequenza. Tali informazioni ed i risultati dell'analisi sono state confrontate con i dati biochimici disponibili.
I dati di partenza di questo lavoro soffrivano di evidenti limitazioni, dovute alla difficoltà dello studio strutturale di proteine di membrana, e di conseguenza i risultati dell'analisi non vogliono fornire un modello accurato della proteina LHC II, ma piuttosto fornire una serie di ipotesi che possano essere d'aiuto nell'interpretazione dei dati disponibili e nella pianificazione di ulteriori esperimenti.
Le seguenti addizioni, poste all'interno del file cvff.frc
di InsightII e Discover (cfr. § C.I.1 e § C.I.2) permettono a
tali software di lavorare con i pigmenti di clorofilla.
Ogni inserzione va posizionata nell'apposita sezione del file suddetto; le sezioni sono qui indicate dal loro nome (come appare nel file del campo di forze) in grassetto.
Le informazioni inserite si riferiscono ai tre nuovi tipi atomici definiti (cfr. § D.III.2) e ne descrivono le caratteristiche (distanze di legame, angoli di legame, massa?).
Per il formato del file cvff.frc si rimanda al manuale di Discover.
#atom_types cvff
2.3 101 np' 14.006700 N 4 Sp2 nitrogen used to simulate interactions of Mg in chlorophylls
2.3 101 np" 14.006700 N 4 Sp2 nitrogen used to simulate interactions of Mg in chlorophylls
2.3 101 Mg' 24.305000 Mg 4 Magnesium - Mg - To simulate tetrapyrrhole of chlorophylls
#equivalence cvff
2.3 101 np' n np' np' np' np'
2.3 101 np" n np" np" np" np"
2.3 101 Mg' Mg' Mg' Mg' Mg' Mg'
#auto_equivalence cvff_auto
2.3 101 np' n np np' n_ np_ n_ np_ n_ np_
2.3 101 np" n np np" n_ np_ n_ np_ n_ np_
2.3 101 Mg' Mg Mg Mg' Mg_ Mg_ Mg_ Mg_ Mg_ Mg_
#quadratic_bond cvff
2.3 101 Mg' np' 2.0500 50.8812
2.3 101 Mg' np" 2.0500 50.8812
2.3 101 c5 np' 1.3800 320.0000
2.3 101 c5 np" 1.3800 320.0000
#quadratic_angle cvff
2.3 101 np" Mg' np" 156.4800 80.2975
2.3 101 np' Mg' np' 156.4800 80.2975
2.3 101 np" Mg' np' 87.6600 128.3911
2.3 101 Mg' np' c5 125.9400 62.3681
2.3 101 Mg' np" c5 125.9400 62.3681
2.3 101 c5 np" c5 106.3000 202.0727
2.3 101 c5 np' c5 106.3000 202.0727
2.3 101 cp c5 np' 120.0000 90.0000
2.3 101 cp c5 np" 120.0000 90.0000
2.3 101 c5 c5 np' 120.0000 90.0000
2.3 101 c5 c5 np" 120.0000 90.0000
#angle-angle-torsion_1 cvff
2.3 101 * Mg' np" * 0.0000 2 180.0000
2.3 101 * Mg' np' * 0.0000 2 180.0000
2.3 101 * c5 np' * 0.0000 2 180.0000
2.3 101 * c5 np" * 0.0000 2 180.0000
2.3 101 * cp np' * 0.0000 2 180.0000
2.3 101 * cp np" * 0.0000 2 180.0000
2.3 101 * np" np' * 0.0000
2.3 101 * cp np' * 0.0000
2.3 101 * cp np" * 0.0000
#out_of_plane cvff
2.3 101 c5 np" c5 Mg' 2.0000 2 180.0000
2.3 101 c5 np' c5 Mg' 2.0000 2 180.0000
#angle-angle cvff
2.3 101 np" Mg' np" np' 0.0000
2.3 101 np" Mg' np' np' 0.0000
2.3 101 np" Mg' np' np" 0.0000
2.3 101 np' Mg' np' np" 0.0000
2.3 101 np' Mg' np" np" 0.0000
2.3 101 np' Mg' np" np' 0.0000
2.3 101 c5 np' Mg' c5 0.0000
2.3 101 c5 np" Mg' c5 0.0000
2.3 101 cp c5 c5 np' 0.0000
2.3 101 cp c5 c5 np" 0.0000
2.3 101 cp c5 np' c5 0.0000
2.3 101 cp c5 np" c5 0.0000
2.3 101 np' c5 cp c5 0.0000
2.3 101 np" c5 cp c5 0.0000
2.3 101 c5 np" c5 Mg' 0.0000
2.3 101 c5 np' c5 Mg' 0.0000
#nonbond(12-6) cvff
2.3 101 Mg' 12833296.0000 3125.83800
#reference 101
Atom types Mg', np' and np" added to simulate chlorophyll
@Author Giuseppe Insana
In questo lavoro di tesi gli aminoacidi vengono sempre
indicati con il loro codice e un numero indicante la loro posizione nella
sequenza della proteina LHC II cristallizzata (LHC II tipo I di pisello).
Alcuni aminoacidi chiave vengono di seguito elencati, con indicate le corrispondenze
di questi con i corrispettivi aminoacidi nelle sequenze delle antenne minori
di Zea mays (CP29, CP26, CP24; in corsivo se non riportanti
analogia di funzione con i corrispettivi di LHC II), come nell'allineamento
della famiglia (cfr. § D.V):
| LHC II
Pisum sativum |
CP 29
Zea mays |
CP 26
Zea mays |
CP 24
Zea mays |
| Glu56 | Phe102 | Glu72 | Ala45 |
| Lys60 | Arg106 | Lys76 | Trp49 |
| Glu63 | Ala109 | Glu79 | Glu52 |
| Glu65 | Glu111 | Glu81 | Asp54 |
| His68 | His114 | His84 | His57 |
| Arg70 | Arg116 | Arg86 | Arg59 |
| Gly78 | Gly124 | Gly94 | Gly67 |
| Pro82 | Val128 | Pro98 | Gly71 |
| Glu83 | Glu129 | Glu99 | Gln72 |
| Arg87 | Gly133 | Lys103 | Gly76 |
| Gln131 | Glu166 | Glu145 | Gln101 |
| Glu139 | Glu174 | Glu153 | Glu109 |
| Arg142 | Arg177 | Arg156 | Arg112 |
| Glu175 | Arg208 | Ile187 | Arg175 |
| Lys177 | Gln210 | Lys189 | Lys177 |
| Lys179 | Ala212 | Lys191 | Ala179 |
| Glu180 | Glu213 | Glu192 | Glu180 |
| Asn183 | His216 | Asn195 | His183 |
| Arg185 | Arg218 | Arg197 | Arg185 |
| Gln197 | Gln230 | Gln209 | Glu197 |
| Lys203 | Lys236 | Glu215 | Lys202 |
| Glu207 | Asn240 | Glu219 | Gly206 |
| Asp211 | Thr244 | Lys223 | Leu210 |
| His212 | His245 | His224 | --- |
Vengono quindi riportati con una descrizione del loro significato.
OVERLAP: 0.01 Å
CUTDIS: 12 Å
SWTDIS: 2 Å
queste tre variabili controllano il cutoff per cui calcolare le interazioni di non legame (un metodo questo per diminuire il costo computazionale di una simulazione). In particolare:
La lista degli atomi vicini, comprendente tutte le coppie di atomi che devono essere considerate durante il calcolo delle interazioni di non legame, è automaticamente ricompilata da Discover ogni qualvolta un atomo si muova più di metà della larghezza della zona di "buffer". Quest'ultima è definita come la zona tra la distanza CUTOFF e quella CUTDIS e contiene gli atomi che potrebbero muoversi abbastanza da rientrare nella zona di calcolo delle interazioni di non legame.
Per il calcolo dell'energia di deformazione delle distanze di legame, durante tutte le simulazioni, il potenziale di Morse (cfr. § C.I.2.1) è stato sostituito da un semplice potenziale armonico proporzionale al quadrato della distanza (in Å) tra atomi:
![]()
dove r è la lunghezza del legame, r0 la lunghezza di riferimento e k2 la costante di forza in kcal/(mol·Å2).
Tale potenziale risulta meno oneroso dal punto di vista computazionale ed inoltre il potenziale di Morse - con derivata molto bassa per valori di distanza elevata - può causare problemi nella simulazione di strutture con gravi distorsioni.
I termini "fuori-diagonale" (cfr. § C.I.2.1) non sono stati utilizzati negli esperimenti di simulated annealing a costanti di forza rilassate perché possono diventare instabili quando la struttura è lontana da un minimo energetico.
Sono stati invece reintrodotti nella successiva fase di minimizzazione.
Il programma Procheck [Laskowski et al. 1993] è stato usato per tale controllo di qualità.
La struttura ricostruita con Maxsprout ha poche deviazioni dalla planarità delle unità peptidiche (angoli diedri w ), a scapito di molte distorsioni nelle distanze e negli angoli di legame dello scheletro polipeptidico.
Dopo la minimizzazione con tethering descritta nel testo (cfr. § D.IV.1.2) tali distorsioni vengono praticamente eliminate (ne rimane solo una a carico della distanza Ca -C nel residuo Thr130) ma con una leggera deviazione dalla planarità per gli angoli w , comunque entro limiti di deviazione standard accettabili.
Allen J.F. (1992). Protein phosphorylation in regulation of photosynthesis. Biochem. Biophys. Acta 1098: 275-335.
Allen K.D., Staehelin L.A. (1992). Biochemical characterization of Photosystem II antenna polypeptides in grana and stroma membranes of spinach. Plant Physiol. 100: 1517-1526.
Altschul S.F., Gish W., Miller W., Myers E.W., Lipman D.J. (1990). Basic local alignment search tool. J. Mol. Biol. 215, 403-410.
Aurora R., Rose G.D. (1998). Helix capping. Protein Science 7, 21-38.
Bairoch A., Apweiler R. (1996). The SWISS-PROT protein sequence data bank and its new supplement TREMBL. Nucl. Acids Res. 24: 21-25.
Barton G.J. (1995). Protein secondary structure prediction. Curr. Opin. Str. Biol. 5: 372-376.
Bassi R., Dainese P. (1992). A supramolecular antenna complex from Photosystem II membranes. Eur. J. Biochem. 204: 317-326.
Bassi R., Pineau B., Dainese P., Marquardt J. (1993). Carotenoid-binding proteins of Photosystem II. Eur. J. Biochem. 212: 297-303.
Bassi R., Rigoni F., Giacometti G.M. (1990). Chlorophyll binding proteins with antenna function in higher plants and green algae. Photochem. Photobiol. 52: 1187-1206.
Bassi R., Sandon D., Croce R. (1997). Novel aspects in chlorophyll a/b binding proteins. Physiol. Plant. 100, 769-779.
Benson D.A., Boguski M., Lipman D.J., Ostell J. (1996). GenBank. Nucl. Acids Res. 24: 1-5.
Bergantino E., Dainese P., Cerovic Z., Sechi S., Bassi R. (1995). A post-translational modification of the PS II subunit CP29 protects maize from photoinhibition. J. Biol. Chem. 270: 8474-8481.
Bernstein F.C., Koetzle T.F., Williams G.J.B., Meyer E.F., Brice M.D., Rodgers J.R., Kennard O., Shimanouchi T., Tasumi M. (1977). The Protein Data Bank: a computer based archival file for macromolecular structures. J Mol. Biol. 112: 535-542.
Biosym technologies (1982). Discover user guide and reference manual. San Diego, CA.
Biosym technologies (1995). Insight II 95.0 Molecular modeling system. User guide. San Diego, CA.
Booth P.J., Paulsen H. (1996). Assembly of light-harvesting chlorophyll a/b complex in vitro. Time-resolved fluorescence measurements. Biochem. 35(16), 5103-5108.
Born M., Oppenheimer J.R. (1927). Zur Quantentheorie der Molekeln. Annal. Physik 84: 457-484.
Bowie J.U., Luthy R., Eisenberg D. (1991). A Method to Identify Protein Sequences That Fold into a Known Three-Dimensional Structure. Science 253: 164-169.
Brejc K.,Ficner R.,Huber R.,Steinbacher S. (1995). Isolation, crystallization, crystal structure analysis and refinement of allophycocyanin from the cyanobacterium Spirulina platensis at 2.3 A resolution. J.Mol.Biol. 249: 424
Chothia C., Lesk A.M. (1986). The relation between the divergence of sequence and structure in proteins. EMBO J. 5: 823-826.
Clarke N.D. (1995). Covariation of residues in the homeodomain sequence family. Protein Science 4: 2269-2278.
Croce R., Breton J., Bassi R. (1996). Conformational changes induced by phosphorylation on the PS II subunit CP29. Biochemistry 35: 11142-11148.
Dainese P., Bassi R. (1991). Subunit stechiometry of the chloroplast Photosystem II antenna system and aggregation state of the component chlorophyll a/b binding proteins. J. Biol. Chem. 266: 8136-8142.
Dauber-Osguthorpe P., Roberts V.A., Osguthorpe D.J., Wolff J., Genest M., Hagler A.T. (1988). Structure and energetics of ligand binding to proteins: E. coli dihydrofolate reductase- trimethoprim, a drug-receptor system. Proteins 4: 31-47.
Dayhoff M. O. (1976). The origin and evolution of protein superfamilies. Fed. Proc. 35: 2132-2138.
Dayhoff M.O. (1978). Atlas of Protein Sequence, Structure. National Biomedical Research Foundation, Washington, D. C., U. S. A.
Deperieux E., Feytmans E. (1992). MATCH-BOX: a fundamentally new algorithm for the simultaneous alignment of several protein sequences. Comput. Appl. Biosci. 8: 501-509.
Eddy S.R. (1995). Multiple alignment using hidden Markov models. In: Rawlings C, et al. (eds). Third International converence on Intelligent Systems for Molecular Biology (ISMB). Menlo Park, CA: AAAI Press, Cambridge, England, pp 114-120.
Ermer O. (1976). Calculation of molecular properties using force fields. Applications in organic chemistry. Structure and Bonding 27: 161-211.
Etzold T., Ulyanov A., Argos P. (1996). SRS: Information retrieval system for molecular biology data banks. Meth. Enzymol. 266: 114-128.
Felsenstein, J. (1989). PHYLIP -- Phylogeny Inference Package (Version 3.2). Cladistics 5: 164-166.
Feng D.-F., Doolittle R.F. (1987). Progressive sequence alignment as a prerequisite to correct phylogenetic trees. J Mol. Evol. 25: 351-360.
Feng D.-F., Johnson M.S., Doolittle R.F. (1985). Aligning amino acid sequences: commonly used methods. J Mol. Evol. 21: 112- 125.
Fogolari F., Esposito G., Viglino P., Cattarinussi S. (1996). Modeling of polypeptide chains as C-alpha chains, C-alpha chains with C-beta, and C-alpha chains with ellipsoidal lateral chains. Biophysical Journal 70: 1183-1197.
Garnier J., Gibrat J.-F., Robson B. (1996). GOR method for predicting protein secondary structure from amino acid sequence. Meth. Enzymol. 266: 540-553.
Gasteiger J., Marsili M. (1980). Iterative partial equalization of orbital electronegativity - A rapid access to atomic charges. Tetrahedron 36, 3219-3288.
George D.G., Hunt L.T., Barker W.C. (1996). PIR-international protein sequence database. Meth. Enzymol. 266: 41-59.
Giuffra E., Cugini C., Croce R., Bassi R. (1996). Reconstitution and pigment-binding properties of recombinant CP29. Eur. J. Biochem. 238: 112-120.
Giuffra E., Cugini D., Croce R., Bassi R. (1996). Reconstitution and pigment-binding properties of recombinant CP29. Eur. J. Biochem. 238, 112-120.
Gobel U., Sander C., Schneider R., Valencia A. (1994). Correlated mutations and residue contacts in proteins. Protein. Struct. Funct. Genet. 18: 309-317.
Gonnet G.H., Cohen M.A., Benner S.A. (1992). Exhaustive matching of the entire protein sequence database. Science 256: 1443-1445.
Green B.R., Pichersky E., Kloppstech K. (1991). Chlorophyll a/b- binding proteins: an extended family. TIBS 16: 181-186.
Gribskov M., Luethy R., Eisenberg D. (1990). Profile analysis. Meth. Enzymol. 183: 146-159.
Hagler A.T., Dauber P., Lifson S. (1979). Consistent force field studies of intermolecular forces in hydrogen bonded crystals. III. The C=OúúúH-O hydrogen bond and the analysis of the energetics and packing of carboxylic acids. J. Am. Chem. Soc. 101: 5131-5141.
Hagler A.T. (1985). Theoretical simulation of conformation, energetics, and dynamics of peptides. Conf. in Biol. & Drug Design, the Peptides 7: 213-299.
Hartley B.S. (1970). Homologies in serine proteases. Phil. Trans. Roy. Soc. London, Ser. B 257: 77-87.
Henikoff S., Henikoff J.G. (1992). Proc. Natl. Acad. Sci. USA 89: 10915-10919
Henikoff S., Henikoff J.G. (1993). Performance evaluation of amino acid substitution matrices. Proteins 17: 49-61.
Henikoff S., Henikoff J.G. (1994). Position-based sequence weights. J. Mol. Biol. 243: 574-578.
Hill T.L. (1960). An introduction to statistical thermodynamics, Dover Publications Inc., N.Y.
Hinze J., Jaff H.H. (1962). Electronegativity. I. Orbital electronegativity of neutral atoms. J. Am. Chem. Soc. 84: 540- 546.
Hinze J., Jaff H.H. (1963). Electronegativity. IV. Orbital electronegativities of the neutral atoms of the periods three A and four A and of positive ions of periods one and two. J. Phys. Chem. 67: 1501-1506.
Hinze J., Whitehead M.A., Jaff H.H. (1962). Electronegativity. II. Bond and orbital electronegativities. J. Am. Chem. Soc. 85: 148-154.
Hobe S., Frster R., Klinger J., Paulsen H. (1995). N-proximal sequence motif in light-harvesting chlorophyll a/b-binding protein is essential for the trimerization of light-harvesting chlorophyll a/b complex. Biochemistry 34: 10224-10228.
Hobe S., Prytulla S., Kühlbrandt W., Paulsen H. (1994). Trimerization and crystallization of reconstituted light- harvesting chlorophyll a/b complex. EMBO Journal 13: 3423-3429.
Hofmann E.,Wrench P.M.,Sharples F.P.,Hiller R.G.,Welte W.,Diederichs K. (1996). Structural basis of light harvesting by carotenoids: peridinin-chlorophyll-protein from Amphidinium carterae. Science 272: 1788
Holm L., Sander C. (1991). Database algorithm for generating protein backbone and side chain co-ordinates from a Ca trace. Application to model building and detection of co-ordinate errors. J. Mol. Biol. 218: 183-194.
Jansson S., Pichersky E., Bassi R., Green B.R., Ikeuchi M., Melis A., Simpson D.J., Spangfort M., Staehelin L.A., Thornber J.P. (1992). A nomenclature for the genes encoding the chlorophyll a/b-binding proteins of higher plants. Plant Mol. Biol. Rep. 10: 242-253.
Jegerschld C., Rutherford A.W., Mattioli T.A., Crimi M., Bassi R. (1999). Calcium binding to the photosystem II subunit CP29. J. Biol. Chem. submitted.
Jones D.T., Taylor W.R., Thornton J.M. (1992). A new approach to protein fold recognition. Nature 358: 86-89.
Kabsch W., Sander C. (1984). On the use of sequence homologies to predict protein structure: Identical pentapeptides can have completely different conformations. Proc. Natl. Acad. Sc., USA 81: 1075-1078.
Kleywegt G.J., Jones T.A. (1998). Databases in protein crystallography. Acta Cryst. D54: 1119-1131.
Koepke J.,Hu X.,Muenke C.,Schulten K.,Michel H. (1996). The crystal structure of the light-harvesting complex II (B800-850) from Rhodospirillum molischianum. Structure (London) 4: 581- 597.
Krogh A., Brown M., Mian I.S., Sjlander K., Haussler D. (1994). Hidden Markov Models in Computational Biology: Applications to Protein Modeling. J. Mol. Biol. 235: 1501-1531.
Kühlbrandt W., Wang D.N., Fujiyoshi Y., (1994). Atomic model of plant light-harvesting complex by electron crystallography. Nature 367: 614-621.
Kuttkat A., Hartmann A., Hobe S., Paulsen H. (1996). The C- terminal domain of light-harvesting chlorophyll-a/b-binding protein is involved in the stabilisation of trimeric light- harvesting complex. Eur. J. Biochem. 242: 288-292.
Laskowski R. A., MacArthur M. W., Moss D. S., Thornton J. M. (1993). PROCHECK: a program to check the stereochemical quality of protein structures. J. Appl. Cryst. 26: 283-291.
Lawrence C.E., Altschul S.F., Boguski M.S., Liu J.S., Neuwald A.F., Wootton J.C. (1993). Detecting Subtle Sequence Signals: A Gibbs Sampling Strategy for Multiple Alignment. Science 262: 208-214.
Lee J.-I., Hwang P.P., Hansen C., Wilson T.H. (1992). Possible salt bridges between transmembrane -helices of the lactose carrier of Escherichia coli. J. Biol. Chem. 267: 20758-20764.
Lipman, D.J., Altschul, S.F., Kececioglu, J.D. (1989). A tool for multiple sequence alignment. Proc. Natl. Acad. Sci. 86:12 4412-4415.
Livingstone C.D., Barton G.J. (1993). Protein sequence alignment: a strategy for the hierarchical analysis of residue conservation. Comput. Appl. Biosci. 9: 745-756.
Maple J.R., Hwang M.J., Stockfisch T.P., Dinur U., Waldman M., Ewig C.S., Hagler A.T. (1994). Derivation of Class II force fields. 1. Methodology and quantum force field for the alkyl functional group and alkane molecules. J. Comput. Chem. 15: 162- 182.
McDermott G., Prince S.M., Freer A., Hawthornthwaite-Lawless A.M., Papiz M.Z., Cogdell R.J., Isaacs N.W. (1995). Crystal structure of an integral membrane light-harvesting complex from photosynthetic bacteria. Nature 6: 517-521.
McGregor M.J., Islam S.A., Sternberg M.J.E. (1987). Analysis of the relationship between side-chain conformation and secondary structure in globular proteins. J. Mol. Biol. 198: 295-310.
McLachlan A.D. (1972). Repeating sequences, gene duplication in proteins. J. Mol. Biol. 64: 417-437.
Michel H.P., Buvinger W.E., Bennet J. (1990). Redox control and sequence specificity of a thylacoid protein kinase. Current research in photosynthesis, Vol II (Baltscheffsky M. ed): 747- 753. Kluwer academic publishers, Dordrecht.
Mulliken R.S. (1934). J. Chem. Phys. 2: 782-793.
Musafia B., Buchner V., Arad D. (1995). Complex salt bridges in proteins: statistical analysis of structure and function. J. Mol. Biol. 254: 761-770.
Nakashima H., Nishikawa K. (1992). The amino acid composition is different between the cytoplasmic, extracellular sides in membrane proteins. FEBS Lett. 303: 141-146.
Needlman S.B., Wunsch C.D. (1970). A general method applicable to the search for similarities in the amino acid sequence of two proteins. J. Mol. Biol. 48: 443-53.
Nether E. (1994). How frequent are correlated changes in families of protein sequences? Proc. Natl. Acad. Sci. USA 91: 98-102.
Nilges M., Clore G.M., Gronenborn A.M. (1988). Determination of three-dimensional structures of proteins from interproton distance data by dynamical simulated annealing from a random array of atoms. FEBS Lett. 239: 129-136.
Nussberger S., Drr K., Wang D.N., Kühlbrandt W. (1993). Lipid- protein interactions in crystals of plant light-harvesting complex. J. Mol. Biol. 234: 347-356.
Overington J., Donnelly D., Johnson M.S., Sali A., Blundell T.L. (1992) Environment-specific amino acid substitution tables: tertiary templates and prediction of protein folds. Prot. Sci. 1: 216-226.
Pagano A., Cinque G., Bassi R. (1998). In vitro reconstitution of the recombinant Photosystem II light-harvesting complex CP24 and its spectroscopic characterization. J. Biol. Chem. 273: 17154-17165.
Pauling L., Yost D.M. (1932). The additivity of the energies of normal covalent bonds. Proc. Natl. Acad. Sci. U.S. 14: 414-416.
Paulsen H., Kuttkat A. (1993). Pigment complexes of light- harvesting chlorophyll a/b-binding protein are stabilized by a segment in the carboxy-terminal hydrophilic domain of the protein. Photochem. Photobiol. 57: 139-142.
Pesaresi P., Sandon D., Giuffra E., Bassi R. (1997). A single point mutation (E166Q) prevents dicyclohexylcarbodiimide binding to the photosystem II subunit CP29. FEBS Letters 402, 151-156.
Peter G.F., Thornber J.P. (1991). Biochemical composition and organization of higher plant Photosystem II light-harvesting pigment-proteins. J. Biol. Chem. 266: 16745-16754.
Pichersky E., Jansson S. (1996). The light-harvesting chlorophyll a/b binding polypeptides and their genes in angiosperm and gymnosperm species. Oxygenic photosynthesis: the light reactions (Ort D.R., Yocum C.F. eds): 493-506. Kluwer academic publishers, Dordrecht.
Pingchiang C. L., Gans P.J., Kallenbach N.R. (1992). Energy contributions of solvent-exposed ion pairs to alpha-helix structure. J. Mol. Biol. 223: 343-350.
Poms R., McCammon J.A. (1990). Mass and step length optimization for the calculation of equilibrium properties by molecular dynamics simulation. Chem. Phys. Letters 166: 425- 428.
Preusser A. (1989). Algorithm 671 - FARB-E-2D: Fill Area with Bicubicson Rectangels - A contour plot program. ACM Transact. on Math. Software 15: 79-89.
Remelli Rosaria (1999). Le basi strutturali della fotosintesi: analisi mutazionale e identificazione dei cromofori nella proteina LHC II. Tesi di laurea.
Risler J.-L., Delorme M.-O., Delacroix H., Henaut A. (1988). Amino acid substitutions in structurally related proteins. A pattern recognition approach. J. Mol. Biol. 204: 1019-1029.
Ros F., Bassi R., Paulsen H. (1998). Pigment-binding properties of the recombinant photosystem II subunit CP26 reconstituted in vitro. Eur. J. Biochem. 253: 653-658.
Rost B. (1996). PHD: predicting one-dimensional protein structure by profile based neural networks. Meth. Enzymol. 266: 525-539.
Rost B. (1997). Protein structures sustain evolutionary drift. Folding & Design 2: S19-S24.
Rost B., Casadio R., Fariselli P. (1996). Topology prediction for helical transmembrane proteins at 86% accuracy. Prot. Sci. 5: in press.
Rost B., Casadio R., Fariselli P., Sander C. (1995). Prediction of helical transmembrane helices at 95% accuracy. Prot. Sci. 4: 521-533.
Rost B., Sander C. (1993). Prediction of protein secondary structure at better than 70% accuracy. J. Mol. Biol. 232: 584- 599.
Rost B., Sander C. (1996). Bridging the protein sequence- structure gap by structure predictions. Annu. Rev. Biophys. Biomol. Struct. 25: 113-136.
Rost B., Sander C., Schneider R. (1993). Progress in protein structure prediction? Trends Biochem. Sci. 18: 120-123.
Rost B., Sander C., Schneider R. (1994). PHD - an automatic mail server for protein secondary structure prediction. CABIOS 10: 53-60.
Russell R.B., Barton G.J. (1992). Multiple Protein sequence alignment From Tertiary Structure Comparison: Assignment of Global and Residue Confidence Levels. Proteins 14: 309-323.
Sander C., Schneider R. (1991). Database of homology-derived structures and the structurally meaning of sequence alignment. Proteins 9: 56-68.
Sandon D., Croce R., Pagano A., Crimi M., Bassi R. (1998). Higher plants light harvesting proteins. Structure and function as revealed by mutation analysis of either protein or chromophore moieties. BBA 1365: 207-214.
Schuler G.D., Altschul S.F., Lipman D.J. (1991). A Workbench for Multiple Alignment Construction and Analysis. Proteins Struct. Funct. Genet. 9: 180-190.
Schulz G.E., Schirmer R.H. (1979). Principles of protein structure. Springer-Verlag Inc, N.Y.
Shindyalov I.N., Kolchanov N.A., Sander C. (1994). Can three- dimensional contacts in protein structures be predicted by analysis of correlated mutations? Protein Eng. 7: 341-348.
Shomer B., Harper R.A.L., Cameron G.N. (1996). Information services of the European Bioinformatics Institute. Meth. Enzymol. 266: 3-27.
Simonetto Roberto (1998). Le basi strutturali della fotosintesi: determinazione dell'orientamento dei cromofori nella proteina antenna CP29. Tesi di laurea.
Simpson D.J., Knoetzel J (1996). Light-harvesting complexes of plants and algae: introduction, survey and nomenclature. Oxygenic photosynthesis: the light reactions (Ort D.R., Yocum C.F. eds): 493-506. Kluwer academic publishers, Dordrecht.
Sipos L., von Heijne G. (1993). Predicting the topology of eukaryotic membrane proteins. Eur. J. Biochem. 213: 1333-1340.
Sistrom W.R., Griffiths M., Stanier T.Y. (1956). The biology of a photosynthetic bacterium which lacks colored carotenoids. Cell. Comp. Physiol. 48: 473-515.
Taylor W.R. (1986). The classification of amino acid conservation. J. Theor Biol. 119:205-218.
Taylor W.R., Hatrick K. (1994). Compensating changes in protein multiple sequence alignments. Protein Eng. 7: 341-348.
Testi M.G., Croce R., Polverino de Laureto P., Bassi R. (1997). A 'CK2' site is reversibly phosphorylated in the PS II subunit CP29. FEBS Lett. 399: 245-250.
Thompson J., Higgins D., Gibson T. (1994). CLUSTAL W: improving the sensitivity of progressive multiple sequence alignment through sequence weighting, position-specific gap penalties nd weight matrix choice. Nucl. Acids Res. 22: 4673-4690.
Tronrud D.E.,Schmid M.F.,Matthews B.W.(1986). Structure and X- ray amino acid sequence of a bacteriochlorophyll A protein from Prosthecochloris aestuarii refined at 1.9 A resolution. J.Mol.Biol. 188:443
Varotto Claudio (1998). Tesi di laurea.
von Heijne G. (1992). Membrane protein structure prediction. J. Mol. Biol. 225: 487-494.
Vriend G. (1990). WHAT IF: a molecular modeling and drug design program. J. Mol. Graph. 8: 52-56.
Walters R.G., Ruban A.V., Horton P. (1994). Higher plant light- harvesting complexes LHCIIa and LHCIIc are bound by dicyclohexylcarbodiimide during inhibition of energy dissipation. Eur. J. Biochem. 226: 1063-1069.
Weiner S.J., Kollman P.A., Case D.A., Singh U.C., Ghio C., Alagona G., Profeta S.Jr., Weiner P. (1984). A new force field for molecular mechanical simulation of nucleic acids and proteins. J. Am. Chem. Soc. 106, 765-784.
Yamamoto H.Y., Bassi R. (1996). Carotenoids: localization and function. Oxygenic photosynthesis: the light reactions (Ort D.R., Yocum C.F. eds): 539-563. Kluwer academic publishers, Dordrecht.
Zuber H., Brunisholz R.A. (1991). Structure and function of antenna polypeptides and chlorophyll-protein complexes: principles and variability. In: H ScheerEditor, Chlorophylls, pp. 627-704. Boca Raton: CRC Press.
 Figura
-4: La molecola di clorofilla
Figura
-4: La molecola di clorofilla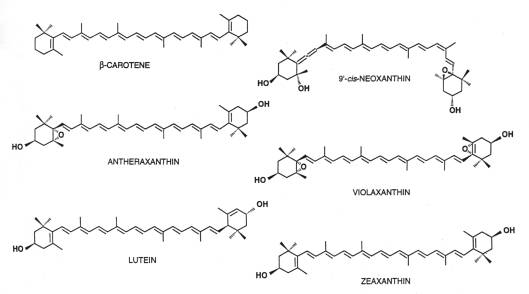 Figura
-6: Struttura di alcuni carotenoidi
Figura
-6: Struttura di alcuni carotenoidi